标题:Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome
日期:2019年11月1日
作者:Víctor J. Carrión, Juan Perez-Jaramillo, Viviane Cordovez, Vittorio Tracanna
单位:荷兰,瓦赫宁根生态研究所,微生物生态中心
摘要
一些土壤显示出显著的抑制植物病原体引起疾病的能力,这种能力归因于与植物相关的微生物群。Carrión等研究枯萎真菌茄枯萎病菌感染甜菜,内生菌(在根中发现的亲密微生物群落)在真菌疾病抑制中的作用。转录分析表明,几种细菌内生菌种会激活生物合成基因簇,从而抑制疾病。这些生物产生抗真菌效应物,包括可消化真菌细胞壁的酶,以及次生代谢产物,包括吩嗪,聚酮化合物和铁载体,可能有助于抗真菌表型。
生活在植物内部的微生物可以促进植物的生长和健康,但是它们的基因组和功能多样性仍然难以捉摸。在这里,宏基因组学和网络推论表明,植物根部的真菌感染在根内富集了几丁质菊科和黄杆菌科,以及几丁质酶基因和编码非核糖体肽合成酶(nonribosomal peptide synthetases,NRPSs)和聚酮化合物合酶(polyketide synthases,PKSs)的各种未知的生物合成基因簇。菌株级别合成的几丁质和黄杆菌的菌群,以持续抑制真菌根部疾病。然后定点诱变表明,以前未鉴定的黄杆菌中的NRPS-PKS基因簇对于内生菌群抑制疾病至关重要。我们的研究结果表明,内生根微生物群具有许多尚不为人所知的功能性状,可以共同保护植物内外。
前言
植物微生物组研究已经积累了大量的测序数据和丰富的信息,表明了许多植物根际、叶际、种子和胚层中不同微生物群落的多样性和丰富度。然而,迄今为止,很少有研究论证微生物菌群对特定植物表型(即植物生长、发育和健康)的影响。因此,在分子和化学层面上许多植物表型与微生物群结构和功能之间在的因果关系仍然未知。本研究旨在探讨植物内生微生物菌群的基因多样性及对真菌感染植株的保护作用。为此,我们整合了包括网络推断和宏基因组学在内的多种方法,以寻找可以抑制Rhizoctonia solani枯萎病(水稻、小麦和甜菜等几种植物根部的真菌病原菌)的内生菌菌群体和功能基因簇。
抑病土壤是一种特殊的生态系统,在这里,由于微生物群落的保护作用,植物才得以存活。各种类型的抑制性土壤都相继报道过,包括真菌、细菌、卵菌和线虫的抑病土壤。抑病性可以通过加热消失,也可以移植到没有抑病作用的土壤中,类似于人类的粪菌移植。
在田间土壤中,通过感病作物的连续栽培过程中集中爆发病害来诱导土壤对病原菌(例如:R.solani)的特异性抑制作用(类似于人类的疫苗)。一旦抑病性形成,如果种植非寄主植物时,这种抑制不表现;但在寄主植物和特定真菌病原体的存在下,抑病作用重新唤醒。这种特异性抑病性土壤一旦形成,便保护之后种植的同种作物和对病原菌产生抵抗性,但是抑病性却在种植其他作物时不表现。
因此,病原菌、寄主植物及其根际微生物群之间的相互作用是特异性抑病性产生和持续的关键因素。我们以前的研究表明,对病原菌 R.solani 有抑制作用的土壤中,发现甜菜根际的几个细菌属paraurkholderia、假单胞菌(pseudomonas)和链霉菌(streptomyces)起着重要的作用。
为了了解那些生活在植物根组织中的微生物(内生菌)在抑病性产生过程中发挥的作用,我们对生长在抑病土壤中的甜菜幼苗的根内进行了宏基因组测序分析;并鉴定了与抑病相关的微生物群落和功能组成;以区分哪些生物合成基因簇(BGCs)在感染过程中上调,然后重组内生菌群;最后进行位点定向突变,检测特异性BGCs是否在抑病过程中起着重要作用。
结果
内生菌群落多样性和网络分析
附图1展示了研究设计:设置甜菜种植在感病(Conducive, C)/抑病(Suppressive, S)土壤和是否接种病原菌R. solani(R)共计四个处理。在抑病土壤中接种病原菌(S+R)的甜菜发病率为15-30%,在感病土壤中接种病原菌(C+R)的甜菜发病率为80%(附图1A)。
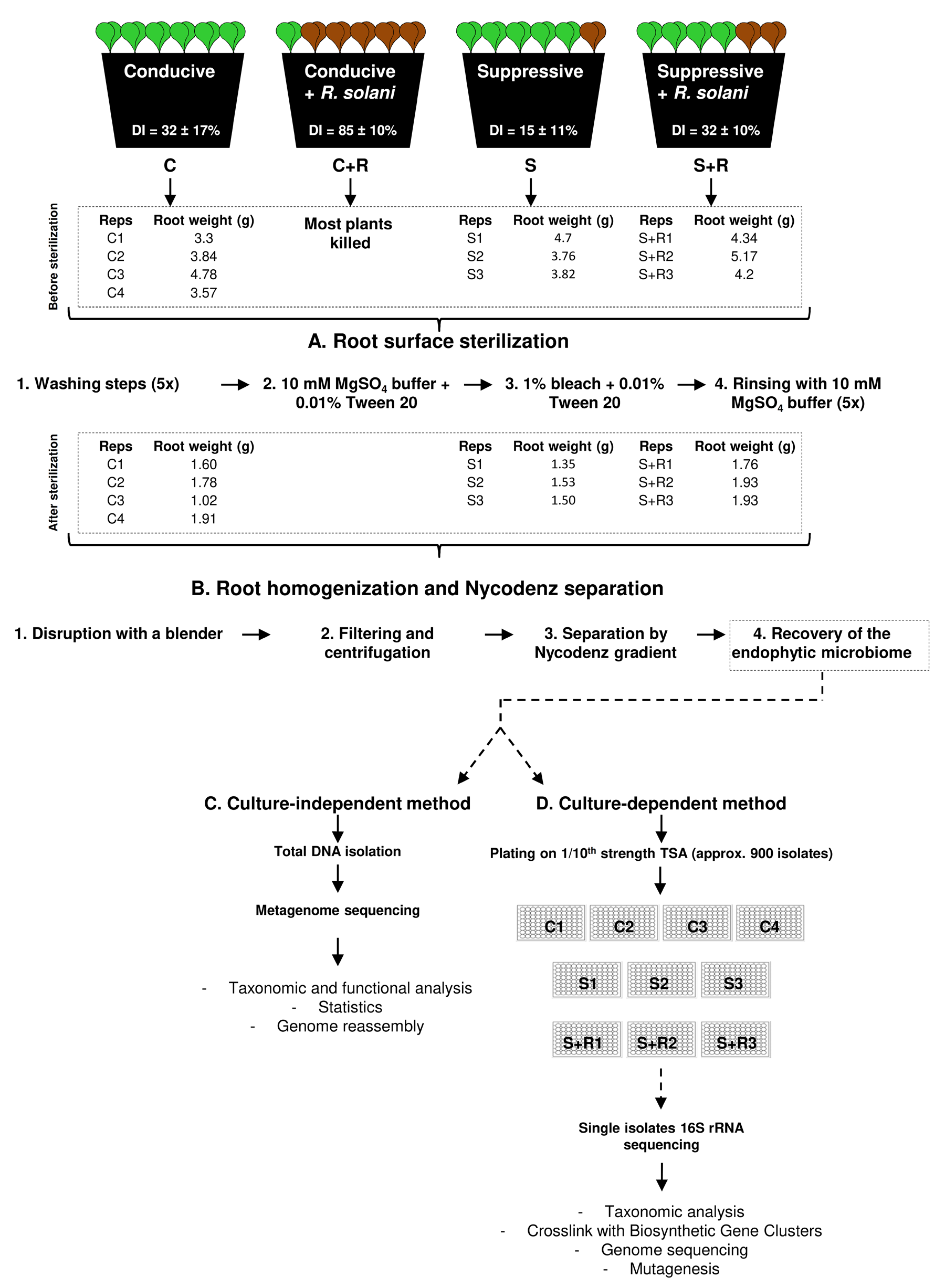
附图1:本研究的实验设计的示意图
因为发病率过高导致没有足够的根系材料用于深入的微生物组学分析。因此作者最终只收集了处理:C,S,和S+R的材料用于下一步测序和生物信息学分析见附图2和附表1/2。
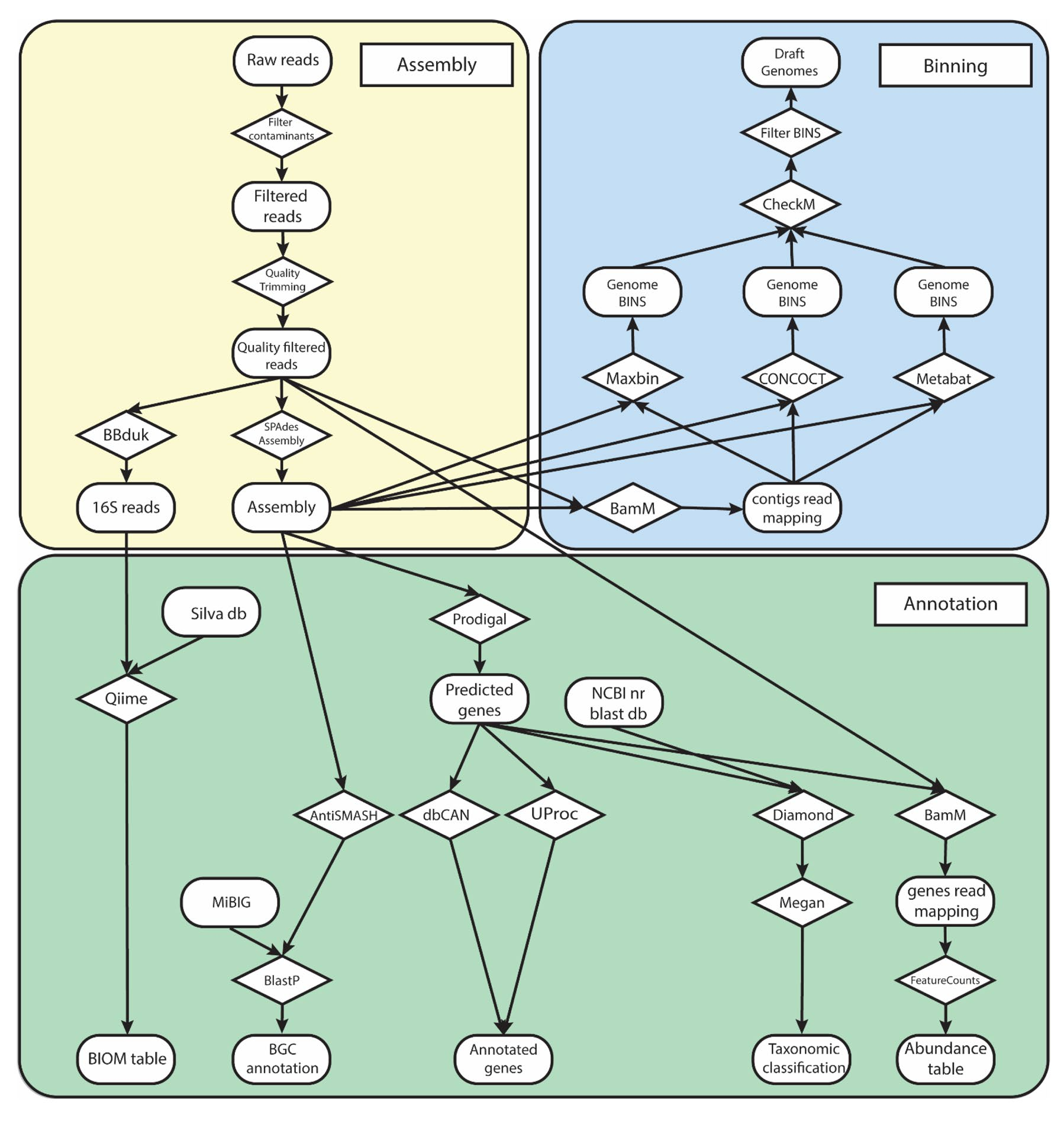
附图2:用于内生菌微生物组的宏基因组学分析流程示意图
宏基因组测序结果显示:76.1%的序列注释为细菌,10.5%的序列注释为真菌,0.0065%的序列被注释为古细菌(附图3A/B)。
对于真菌序列限制性主坐标分析(CPCoA)表明内生真菌在三组之间存在显著差异((PERMANOVA), P < 0.05])(附图4A),这是由于在S+R组大量接种了病原真菌R. solani导致的(附图4B/C)。
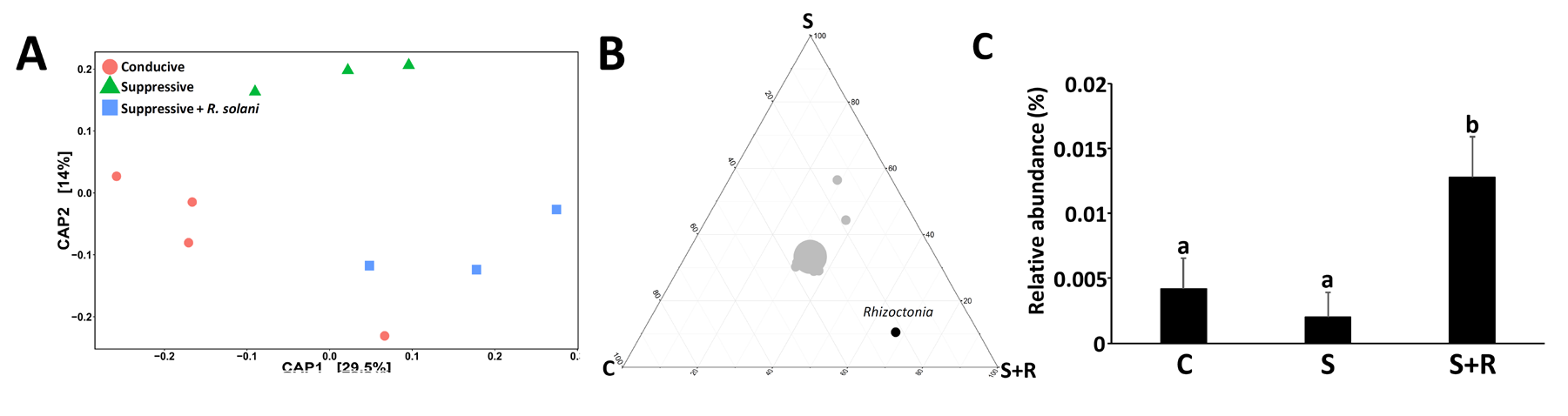
附图4:从宏基因组中提取的真菌reads的概述和组成
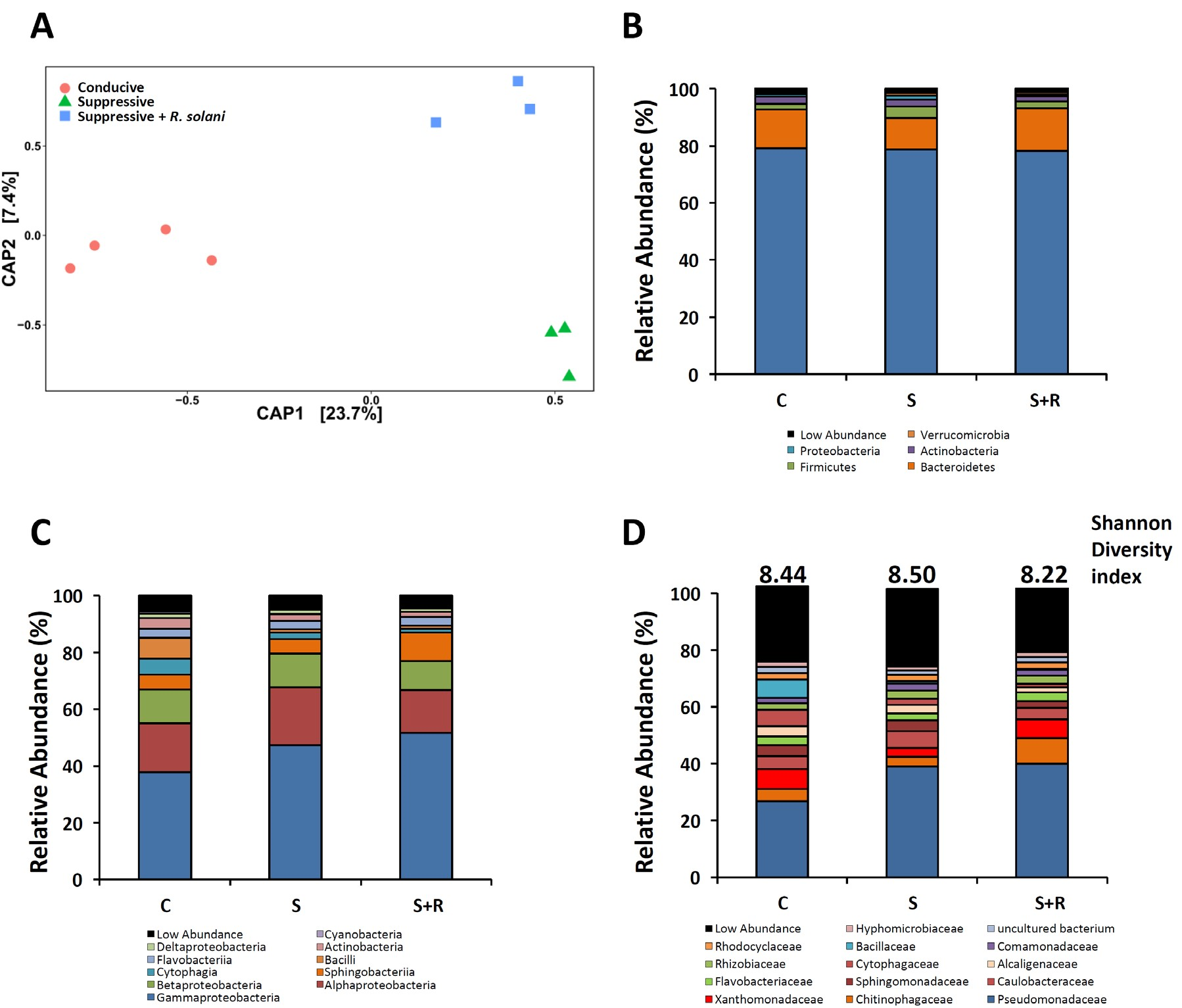
附图5:甜菜内生微生物群的β多样性和α多样性
A: CAP分析展示Beta多样性(可解释23%的总体差异);
B-D: 展示门、纲、科水平各组中的相对丰度,其中D图上展示香农多样性指数且无显著差异。
共线性网络分析表明S+R组内生微生物群落复杂度更高(附表3),相关研究也表明连接性比较高的网络往往出现在微生物群落面对逆境的时候,比如病原菌入侵。有意思的是在S+R网络中80%的连接点都属于Chitinophaga, Flavobacterium和Pseudomonas这三个科。当将属于Bacteroidetes的序列去除,三个分组的差异就难以区分。这再次暗示了Bacteroidetes中的Chitinophaga和Flavobacterium与抑病相关。
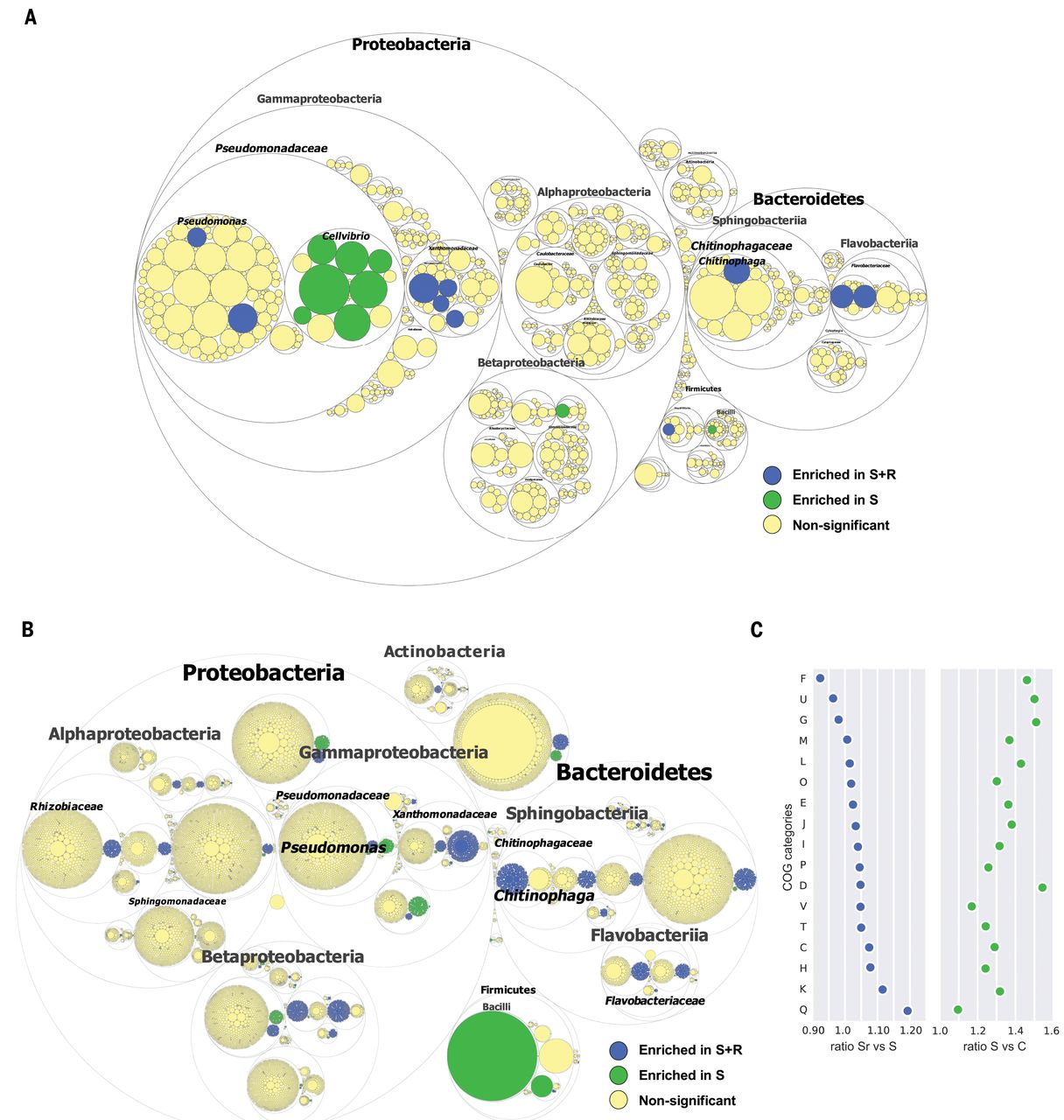
图1. 生长在S和S+R两个土壤中的植物内生不同菌群丰度差异
A:通过宏基因组提取16S rRNA基因序列注释细菌群落并统计不同门类细菌差异,最大的圈代表门水平,逐渐缩小的圈按照梯度分别代表纲,科,属;
B:基于宏基因组序列注释功能和物种相关功能差异。最小的圈代表COG功能单位。圆圈大小代表了不同物种或者功能的平均丰度。S组富集的物种或者功能使用绿色标记,S+R组富集的使用蓝色标记。不显著的物种或者功能使用黄色标记。
C:左边散点图描绘了全部S+R组属于拟杆菌门的全部基因/S组对应基因的丰度比值,右边散点图是S组属于拟杆菌门的全部基因/C组对应基因的丰度比值。散点图从上到下排序顺序按照S+R/S比值。每个COG类别缩写对应如下:C:能量代谢;D:细胞周期,细胞分裂和染色体分区;E:氨基酸运输和代谢;F: 核甘酸转运和代谢;G:碳水化合物的运输和代谢;H:辅酶的运输和代谢;I:脂质的运输与代谢;J:翻译,核糖体结构;K:转录;L:复制,重组和修复;M:细胞壁,细胞膜和细胞被膜的生物发生;O:翻译后修饰,蛋白质转换;P:无机离子的运输和代谢;Q:次生代谢产物的生物合成,转运和分解代谢;T:信号转导机制;U:细胞内运输,分泌和囊泡运输;V:防御机制。
内生菌群落功能多样性
从宏基因组测序数据中得到50%-70%的序列都被注释到了功能(附图3C-E)。通过对其他基因的注释显示有56,175条与微生物类群相关的功能,其中402个功能是显著在S组中显著富集的。在S+R组发现部分基因上调超过10倍,这些基因属于“碳水化合物转运和代谢”和“信号转导机制“。同时发现与这些基因高度相关的微生物门类均在S+R中显著增加,包括:Chitinophagaceae和Flavobacteriaceae (Bacteroidetes); Pseudomonadaceae和 Xanthomonadaceae (Gammaproteobacteria); Hyphomicrobiaceae和Rhizobiaceae (Alpha-proteobacteria); 和 Burkholderiaceae (Betaproteobacteria) (图1B/C,附图9A)。
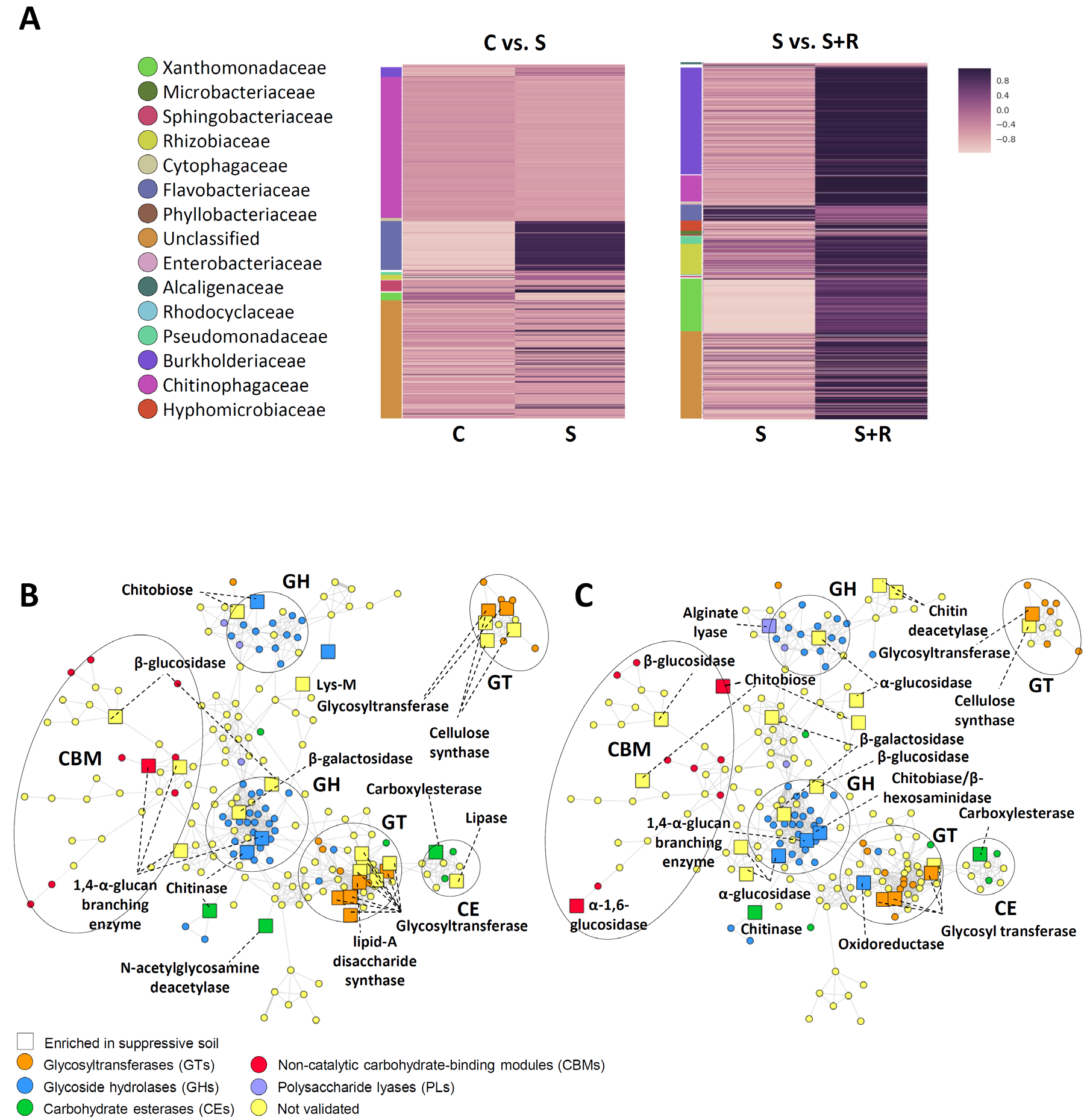
附图9:内生微生物群中碳水化合物活性酶的多样性和分布
A:热图展示各组中显著差异的基因,按物种来源注释分类;
B:碳水化合物代谢相关的HMM结构域与基因间的相似网络。
其中大部分增加的基因(3138/4443)被注释为与Chitinophagaceae 和Flavobacteriaceae(图1B和附图9A)相关。当我们选择更严格的阈值定义上调基因时,发现有461个上调基因,这些基因大部分都与Chitinophagaceae 和Flavobacteriaceae相关。仅对Bacteroidetes相关的基因做差异分析,显示了S+R组和S组间按COG功能注释为Q类(次级代谢产物生物合成、转运和分解代谢)的基因差异最大;而S组和C组之间G类(碳水化合物转运和代谢)基因的差异最大(图1C)。
为了更详细的了解COG的G类和Q类功能的细节,分别查找了碳水化合物活性酶数据库(CAZymes)和次级代谢产物生物合成基因簇数据库antiSMASH。通过dbCAN, 我们注释得到1822个基因主要属于一下功能分类模块:糖苷羟化酶、糖基转移酶、多糖裂解酶和糖酯酶结构域以及非催化性糖结合模块。这些结构域与进化相关并且参与相关功能,所以我们使用hhsearch算法计算蛋白保守结构域的相似性。从而发现S+R组内生菌群中有更多的糖苷水解酶和糖基转移酶与抑病相关(图2A和附图9B/C) 。与S组相比,S+R组有三类内生菌在CAZyme酶注释上显著不同(FDR < 0.1;图2A和附图9A/B)。此外,我们发现Chitinophagaceae含有一些与真菌细胞壁降解的相关酶类。比如:几丁质酶、β-葡聚糖酶和内葡聚糖酶等。注释为Burkholderiaceae 和Xanthomonadaceae科(附图9B/C)的菌有两个几丁质酶结构域和三个参与几丁质降解的蛋白酶。这五个结构域在这三种菌是共有的。表明了这些富集的内生菌在同一种功能上的冗余。
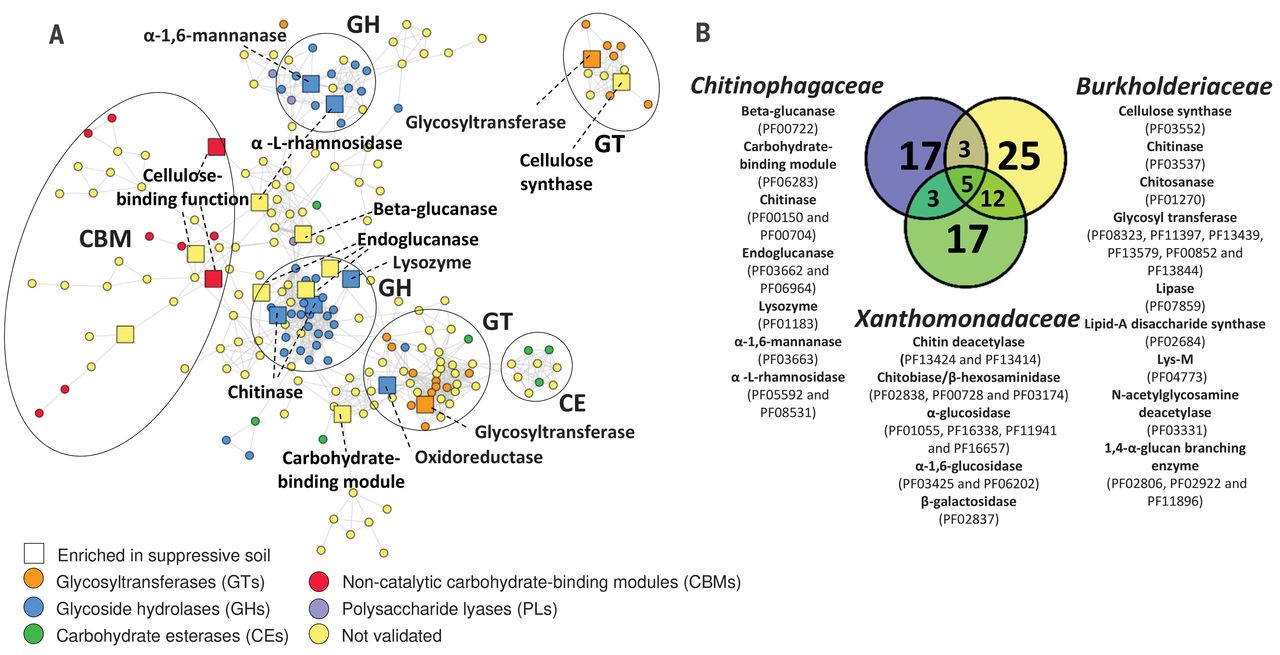
图2:内生菌群碳水化合物相关活性酶的多样性和分布
A:与碳水化合物酶类相关的已知蛋白和预测蛋白结构域的相似性网络。使用网络展示不同蛋白结构域之间的距离和相关性。使用CAZymes对生长在抑病土壤S组或接种病原菌的抑病土壤S+R组的植物内生菌群中注释,一共得到1822个基因。节点分为五大类:糖苷水解酶(GH,蓝色);糖基转移酶(GT,橙色);多糖裂解酶(PL,紫色);糖脂酶(CE,绿色)和非催化碳水化合物结合模块(CBM,红色)。位置结构域或者功能尚未验证的蛋白结构域标记为黄色。方形节点代表物种注释为Chitinophagaceae的相关酶在S+R组中显著高于S组。在S+R中过表达的并在分类学水平上属于Burkholderiaceae和Xanthomonadaceae的相关酶类展示在附图9B/C中。
B:展示标记的三种富集在S+R组的科水平细菌的CAZymes注释数量。韦恩图黄色代表Burkholderiaceae (黄);蓝色代表 Chitinophagaceae (蓝);绿色代表Xanthomonadaceae (绿)。韦恩图标记了每个酶对应Pfam数据库ID。韦恩图展示和每个物种共有的和特有的相关结构域数量。
细菌基因组中包含了大量的BGCs,但是大部分都没有备注到已有的模块和功能。通过antiSMASH分析次级代谢表明730个生物合成基因簇(BGCs)与非核糖体肽、聚酮、萜类、芳基多烯、核糖体合成和翻译后修饰肽(RIPS),膦酸盐、吩嗪和铁载体相关。在这些730个BGCs中已经有12个在先前的研究中报道过,并且物质化学结构也被阐明(附图11和附表5)。其中包括两个脂肽类抗生素thanamycin和brabantamide,并且相关产品也用在土壤中来抑制病原菌Rhizoctonia。其他718个BGCs注释目前还不够成熟,其中117个BGCs显著在S+R组中过表达并且其中34个BGCs属于Bacteroidetes(图3A-F和附图10-12)。值得注意的是这34个BGCs中并不包括先前已经在根际鉴定过的thanamycin和brabantamide。在这过表达的117个基因中有10个NRPS基因簇属于Bacteroidetes,并且通过antiSMASH数据库发现这些都和MIBiG中注释到的基因簇不匹配。
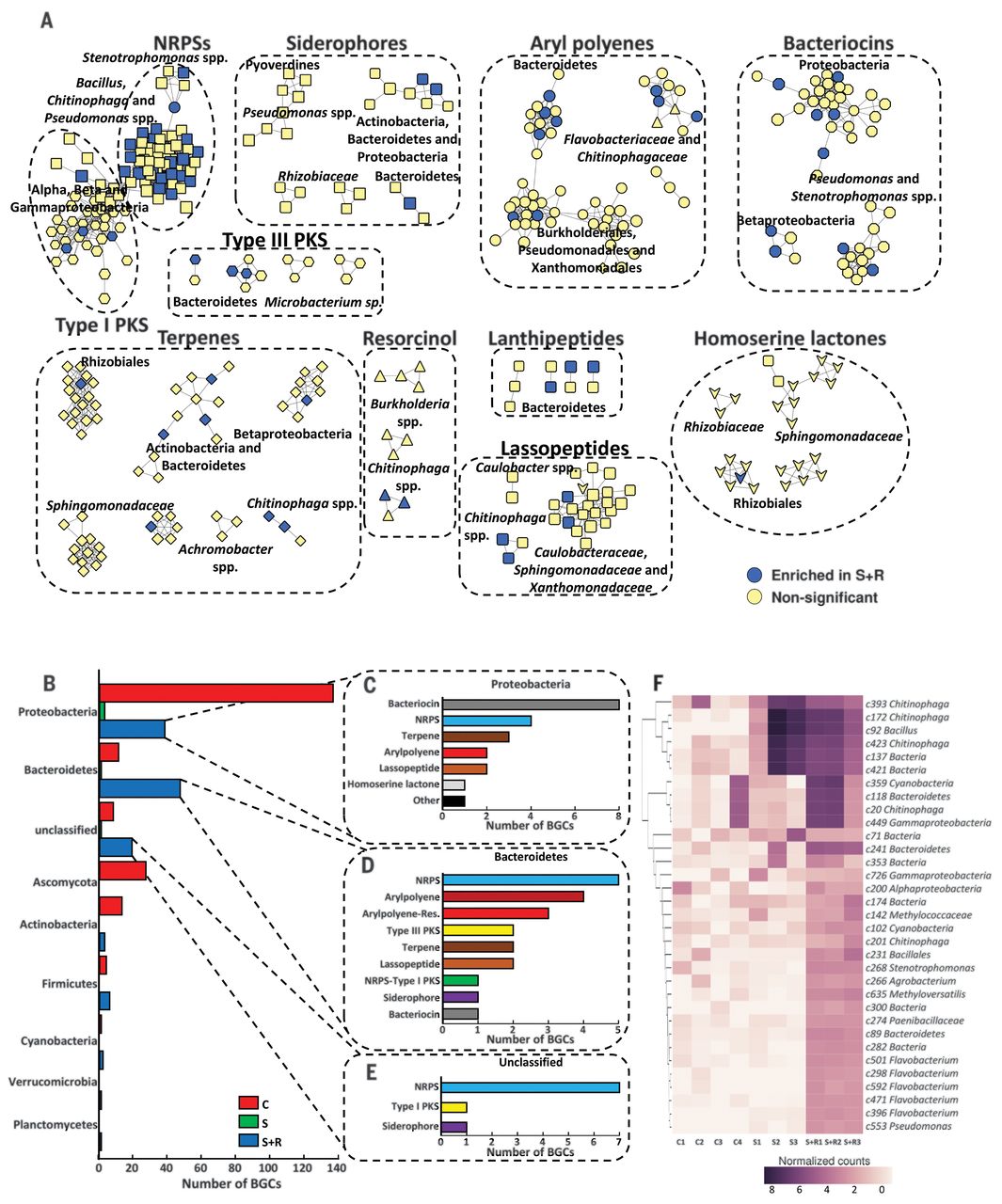
图3:内生微生物群中生物合成基因簇的多样性和分布
从头组装内生菌基因组
从antiSMASH数据库中鉴定得到的730个BGCs中,有157个包含在组装成的25个基因组(MAGs)中(附图13/14和附表6)。MAGs、管家基因和鉴定的BGCs随后被用于生成转录组分析的特异性引物集,并将BGCs与细菌内生菌收集的分离物联系起来。
最初收集的935株细菌内生菌(图S1)通过16S rRNA测序(图S15、A、B和表S7)进行了分类鉴定,发现8个不同的属,主要以Bacteroidetes和Gammaproteobacteria为代表。虽然聚合酶链反应筛选没有检测到与几丁质噬菌或假单胞菌相关的BCGs(表S8),但在S+R条件下获得的内生黄杆菌分离物中发现了4个BCGs(BGC298、BGC396、BGC471和BGC592)。其中3个编码NRPS(BGC396、BGC471和BGC592),第4个编码NRPS-聚酮合成酶(PKS)杂交基因簇(BGC298,图4A)。类似的方法证实了在S+R条件下获得的三株内生食几丁菌分离株中存在糖基水解酶(GH18)基因(图2A)。随后的体外实验表明,这三种食几丁质菌也具有细胞外溶几丁质活性。
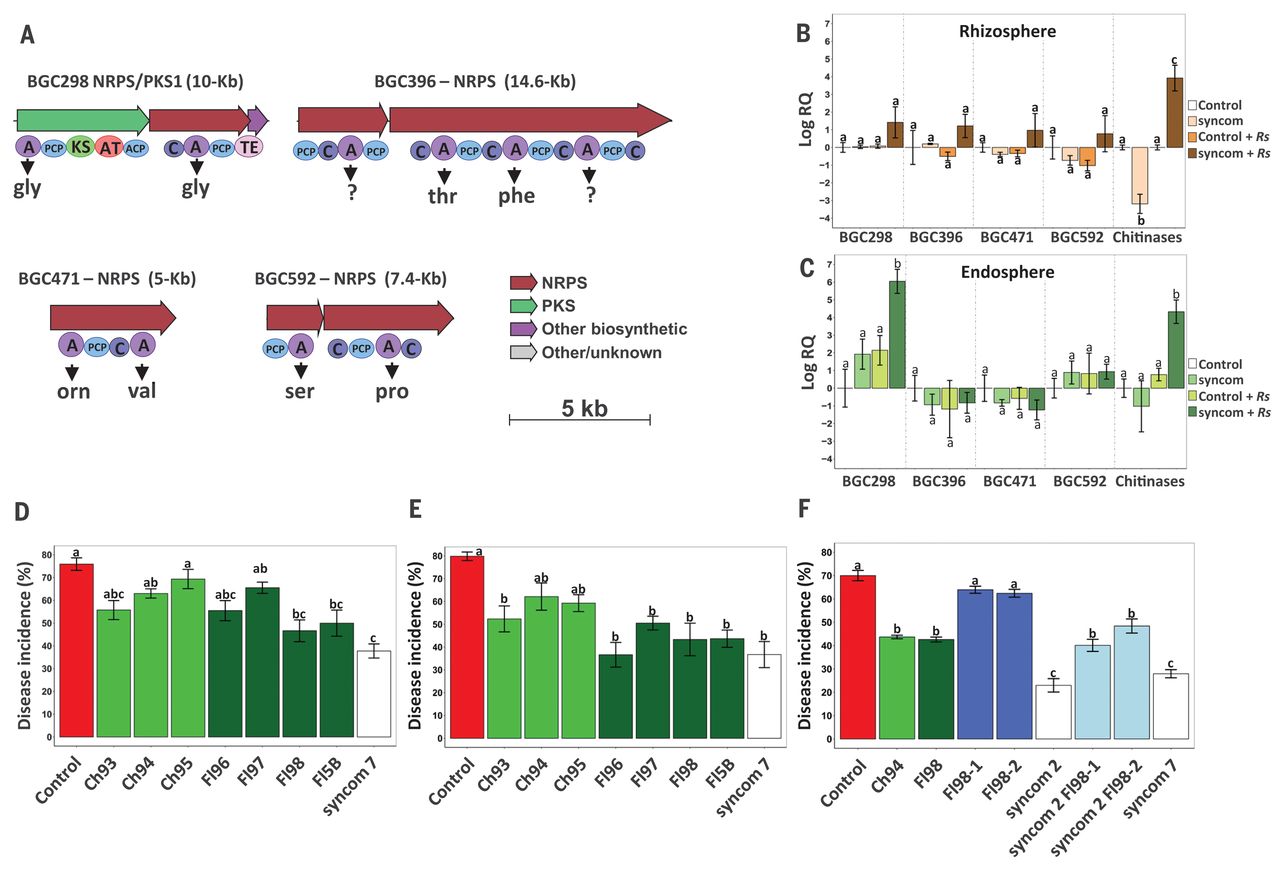
图4:疾病抑制联合体的转录和功能分析
随后对3株食几丁菌和4株黄杆菌的基因组测序显示,每个属内的分离株相似性>99 %。(图S15C、S16A及表S9)。分离的基因组还聚集了分配给每个属的MAG(图S15, B和C),证实它们对应于微生物组中丰富的分类群。在与拟杆菌门分离物的全基因组序列比较的基础上,对于关键的BCGs,没有发现宏基因组错误组装的迹象(图S16BC,S17A-C)。
疾病抑制联合体的重建和功能分析
作者选择了7个已测序的拟杆菌门菌株进行根定植试验和BCG-转录分析。所有菌株都在甜菜幼苗的根际和根内圈定植(图S18和S19)。转录分析显示,几丁质酶在接种真菌病原体的根际和内球区室定植的菌群中表达显著(P < 0.05)升高(图4BC和图S20)。在四个黄杆菌基因簇中,当植物根系受到真菌病原菌R. solani侵染时,BGC298在内圈的表达量显著高于根际(图4C)。该BGC在所有四个黄杆菌基因组和MAG中一致组装(图S16B),并且与MIBiG中已知的BGC不匹配(图S17)。
黄杆菌和食几丁质菌在抑制病害土壤中生长的植物功能网络中的中心位置,它们在内球圈定殖的能力,以及真菌病原菌诱导合成联盟中BGC298和几丁质酶基因的表达,表明黄杆菌和食几丁质菌在病害抑制中起作用。为了验证这一假设,三个独立的生物测定表明,几丁菌和黄杆菌的联合体比单个成员对真菌根感染具有显著和一致的保护作用(图4D-F和图S21A-C)。即使单个分离株对疾病的益处很小,联合体也总是显示出更大程度的保护作用。重组植株表型的明显“最小”联合体由一个食几丁菌分离物和一个黄杆菌分离物(syncom-2)组成,该联合体显示出与7个成员联合体相同的疾病抑制水平(图4f)。
为了证实黄杆菌BGC298在疾病抑制活性中的作用,作者开发了一个SpyCas9介导的系统,用于在黄杆菌98中引入双链DNA断裂。我们获得了两个独立的BGC298突变体(图S22A-D,表S10-12),通过特定引物的Sanger测序验证了PKS基因缺失(图S22D)。这两个突变体在根际和内圈的定殖程度与野生型黄杆菌98单独或与Chitinophaga sp. 94一起引入时相同(表S13)。当两个独立的BGC298突变体在疾病生物测定中进行测试时,突变体降低了黄杆菌98单独和与食几丁菌分离物联合处理时的疾病抑制效果(图4F)。
总结
在我们之前关于土壤抑制真菌根病的研究中,我们发现根际细菌是第一道防线。如果病原体突破这第一道防线,它将遇到植物的基础和诱导防御机制。在这里,我们表明在病原体入侵植物根系的第二阶段,内生微生物组可以提供额外的保护层。我们的实验表明,当病原体入侵时,噬几丁质菌科(Chitinophagaceae)和黄杆菌科(Flavobacteriaceae)的成员在植物根部内富集,并表现出与真菌细胞壁降解相关的酶活性增强,以及由NRPSs和PKs编码的次生代谢物生物合成。从宏基因组序列中重新组装25个细菌基因组后,够重建一个提供疾病保护的黄杆菌和几丁菌合成群落(syncom)。定点突变进一步证实了黄杆菌中BGC298对这种表型的贡献。这两种细菌在根组织中的位置以及它们如何在分子水平上在内球层中相互作用尚不清楚。可能是几丁质酶产生的壳寡糖诱导了黄杆菌BGC298的表达。BGC298编码的代谢物是否具有直接的抗真菌活性或作为其他保护性性状的调节剂尚不清楚。另一个考虑因素是,该联合体可能通过诱导根部的局部或系统性抗性而产生间接影响。本研究的结果突出了内生根微生物组中未知的微生物属和功能性状的财富。采用宏基因组引导的分析和网络推断成功地确定了微生物群落的分类群和功能,从而获得特定的微生物组相关植物表型。