📅 官方地址:https://github.com/molevol-ub/bitacora
🔬 教程版本:BITACORA 1.4
📋 主要功能
- 基因家族识别与注释优化
- 纠正常见注释错误: 自动修正错误基因模型(如跨基因融合转录本、部分外显子缺失或错误剪接位点)。
- 发现新基因家族成员: 通过整合序列相似性搜索工具(如BLAST、HMMER),直接从基因组DNA中识别未注释的基因家族拷贝。
- 非模式生物优化的流程
- 针对缺乏高质量参考基因组的物种(如新测序的非模式生物),提高基因模型的完整性和准确性。
- 数据整合与输出
- 生成标准化文件:
- GFF 文件: 包含修正后的基因模型和新发现的基因家族拷贝结构信息。
- FASTA 文件: 提供所有预测的蛋白质序列,便于后续功能注释和分析。
- 兼容主流基因组注释编辑器(如Apollo、Geneious),简化人工校验和下游分析。
- 生成标准化文件:
(以上由Deepseek生成)
🔧 依赖环境:
Perl
# https://learn.perl.org/installing/
BLAST
# ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
HMMER
# http://hmmer.org/
GeMoMa
# https://www.jstacs.de/index.php/GeMoMa
# BITACORA软件压缩包中默认包含了V1.7.1版本的GeMoMa
🧷 安装方式:
# 下载解压即可
wet https://github.com/molevol-ub/bitacora/archive/refs/tags/BITACORA-v1.4.2.tar.gz
tar zxf BITACORA-v1.4.2.tar.gz
mv bitacora-BITACORA-v1.4.2/ bitacora-v1.4.2
cd bitacora-v1.4.2
unzip GeMoMa-1.7.1.zip && rm GeMoMa-1.7.1.zip __MACOSX/ -rf
chmod 755 runBITACORA*
🚩 主要参数介绍:
bitacora-v1.4.2目录下runBITACORA.sh中有EXPORT EXECUTABLES TO PATH、PREPARE THE DATA、EDITABLE PARAMETERS、HOW TO RUN、PIPELINE - CODE五个部分。
# EXPORT EXECUTABLES TO PATH
# 添加HMMER和BLAST程序、BITACORA脚本(Scripts)、GeMoMa路径
# PREPARE THE DATA
# NAME 设置待分析物种的名称
# GENOME 待分析物种的基因组数据,.fasta文件
# GFFFILE 待分析物种的基因组的GFF3或GTF文件。如果是来自NCBI的GFF文件,则需要使用“reformat_ncbi_gff.pl”脚本进行转换(位于/Scripts/Tools目录下)
# PROTFILE GFF预测蛋白质的fasta文件
# QUERYDIR 包含FPDB数据库的文件夹(包括一个命名为YOURFPDB_db.fasta的fasta文件和命名为YOURFPDB_db.hmm的HMM文件);可以在文件夹中包含多个FPDB以进行搜索
!!![NOTE: mRNA or transcript, and CDS are mandatory fields]
使用细菌基因组的GFF文件,用reformat_ncbi_gff.pl转换后得到的文件为空。
📰 软件测试
一个运行BITACORA的示例可以在Example文件夹中找到。首先,解压缩Example_files.zip文件以获取BITACORA所需的文件。在此示例中,将在果蝇(Drosophila melanogaster)的2R染色体中搜索两个与化学感应相关的基因家族:气味受体(ORs)和CD36-SNMP基因家族。GFF3和蛋白质文件是从原始注释中修改的,删除了某些基因模型,以便BITACORA能够识别未注释的新基因。</span>
📋 测试数据:
# bitacora-v1.4.2/Example文件中的数据,包括Files目录下果蝇(Deml_genecounts)基因组fasta文件、GFF文件和蛋白质fasta文件,还有DB中CD36-SNMP和OR的FPDB数据库
# 在Deml_genecounts中查询CD36-SNMP和OR家族基因
📑 测试代码:
将主目录下的runBITACORA.sh拷贝到Example目录下,正确配置后直接运行
bash runBITACORA_v1.4_demo.sh
📣 结果解释:
# 三个汇总的表格文件:
Deml_genecounts_annotated_and_genomic_proteins.txt # 基于注释和识别未注释的蛋白质汇总
Deml_genecounts_annotated_proteins.txt
Deml_genecounts_summary.txt
# 不同FPDB家族会生成不同的目录结果,包括以下主要文件:
CD36-SNMP_genomic_and_annotated_genes.gff3
CD36-SNMP_genomic_and_annotated_genes_nr.gff3
CD36-SNMP_genomic_and_annotated_proteins_trimmed.fasta
CD36-SNMP_genomic_and_annotated_proteins_trimmed_idseqsclustered.fasta
CD36-SNMP_genomic_and_annotated_proteins_trimmed_idseqsclustered.gff3
CD36-SNMP_genomic_and_annotated_proteins_trimmed_idseqsclustered.gff3_overlapping_genes.txt
CD36-SNMP_genomic_and_annotated_proteins_trimmed_idseqsclustered_table.txt
CD36-SNMP_genomic_and_annotated_proteins_trimmed_nr.fasta
CD36-SNMPtblastn_parsed_list_genomic_positions.bed
CD36-SNMPtblastn_parsed_list_genomic_positions_nogff_filtered.bed
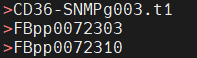
如:从Deml中注释到了2个CD36-SNMP家族蛋白(FBpp0072303和FBpp0072310),并发现了一个新的CD36-SNMP家族蛋白成员CD36-SNMPg003.t1
cat CD36-SNMP_genomic_and_annotated_proteins_trimmed_idseqsclustered.fasta|grep ">"
runBITACORA_genome_mode
在使用runBITACORA.sh处理Prokka生成的.gff和.faa文件时会出现报错信息,可以通过使用genome模式的runBITACORA_genome_mode.sh命令从基因组层面进行数据处理,可顺利运行。
🏷️ 标签:bioinfomatics/phyloinfomatics,gene families,gene structure and function,genomics,molecular evolution,structural annotation,transcriptomics
📅 创建:2025-02-28
🔗 原文:查看文献