标题:Microbial Diversity Biased Estimation Caused by Intragenomic Heterogeneity and Interspecific Conservation of 16S rRNA Genes
日期:2023年4月27日
作者:周宁一
单位:武汉病毒研究所
本研究是继2013年的一篇题为“Intragenomic Heterogeneity of 16S rRNA Genes Causes Overestimation of Prokaryotic Diversity”的研究。以下是该文章(2013)的相关信息:
自从Carl Woese应用16S rRNA基因来确定原核生物的亲缘关系,这个方法已成为原核生物系统发育和分子生态学研究中公认的经典标准。然而,该基因在原核生物内往往同时存在多个拷贝,而且拷贝之间的基因序列并不完全一致。因此在原核生物生态学研究中,基于16S rRNA基因的菌群多样性分析会引起一定程度的高估。该组从目前2013个测序的原核生物基因组中,对16S rRNA基因的拷贝数及基因组内部异质性做了详细的分析研究,发现细菌基因组中包含1-15个拷贝,古菌基因组中包含1-4个拷贝。在952个基因组(隶属于585个种)内发现16S rRNA基因存在异质性。并发现16S rRNA基因不同区域存在不同程度的异质性,因而利用不同区域的序列进行基于焦磷酸测序的分子生态学研究会造成不同程度的多样性高估。本研究指出对于经常用来进行焦磷酸测序的区域中,V4-V5区域显示了最低的高估程度(约为3.0%),而V6区域的高估程度最高(约为13%)。因此在原核生物分子生态学研究中16S rRNA基因的V4-V5区域更适合作为焦磷酸测序的目的片段。这是迄今为止最全面的关于16S rRNA基因的基因组内差异的研究,本研究不但提供了在针对不同的16S rRNA基因区段时不同丰度的高估程度,而且提出了可利用V4-V5区来降低这一高估。
摘要
16S rRNA基因已被广泛用作分子标记,以探索各种环境中的进化关系和微生物组成。尽管其方便且普遍存在,但不可避免存在局限性。可变拷贝数、基因组内异质性和低分类学分辨率导致了估计微生物多样性的偏差。本文分析了24248个完整的原核生物基因组,表明细菌中16S rRNA基因拷贝数从1到37不等,古菌中从1到5不等,并且观察到60%的原核生物基因组存在基因组内异质性,其中大多数低于1%。对使用全长或部分16S rRNA基因时,基因组内变异引起的微生物多样性的高估和种间保守引起的低估进行了计算。结果表明,在100%阈值下,使用全长基因时微生物多样性可能被高估多达156.5%。基于V4至V5区域的分析引入了最低的过高估计率(4.4%),但在97%阈值下显示出略低于其他可变区域的物种分辨率。对于不同的可变区域,提出了使用适当的阈值而不是典型值97%,以最大限度地减少将单个基因组拆分为多个物种并将不同物种聚集成同一簇的风险。这项研究不仅更新了当前可用原核生物基因组的16S rRNA基因拷贝数和基因组内变异信息,还阐明了使用定量数据估计原核生物多样性的偏差,为选择扩增区域和聚类阈值提供了参考。
结果
本研究于2021年11月从美国国家生物技术信息中心(NCBI)的完整基因组数据库中获得了属于6889个不同物种的24248个完整基因组(399个古细菌,23849个细菌;基因组的详细信息见补充材料中的文件S1)。从每个基因组中成功检索到16S rRNA基因序列。这6889种物种来自46个不同的门(表1),其中最丰富的是变形菌门(3198种),其次是放线菌门(1172种),厚壁菌门(1039种),拟杆菌门(518种),广古菌门(217种),蓝细菌门(159种)和乳杆菌门(137种),其余39个门中每个门代表的物种都少于100种。在进行基因组分类数据库(GTDB)分类时,24248个基因组中有24037个被分类学鉴定到物种水平(6265个GTDB物种),其余基因组未被分类到特定物种。由于使用了NCBI分类法和GTDB分类法,除非另有说明,本研究中提到的分类名称均指NCBI分类法。
古菌和细菌中16S rRNA基因拷贝数
古菌16S rRNA基因拷贝数在1-5之间,细菌在1-37之间(图1)。在Tumebacillus avium AR23208中观察到的最大拷贝数为37,远远大于先前发表的值(15个拷贝)。只有8%的细菌基因组含有单个16S rRNA基因,而超过一半的古菌基因组含有一个拷贝。具有7个拷贝的细菌(4465个基因组)最丰富,拷贝数大于15的相对较少,共有31个基因组。细菌的平均拷贝数为5.36,明显高于古菌(1.76)。总共鉴定到127003个16S rRNA基因拷贝,平均每个原核生物基因组5.2个拷贝。
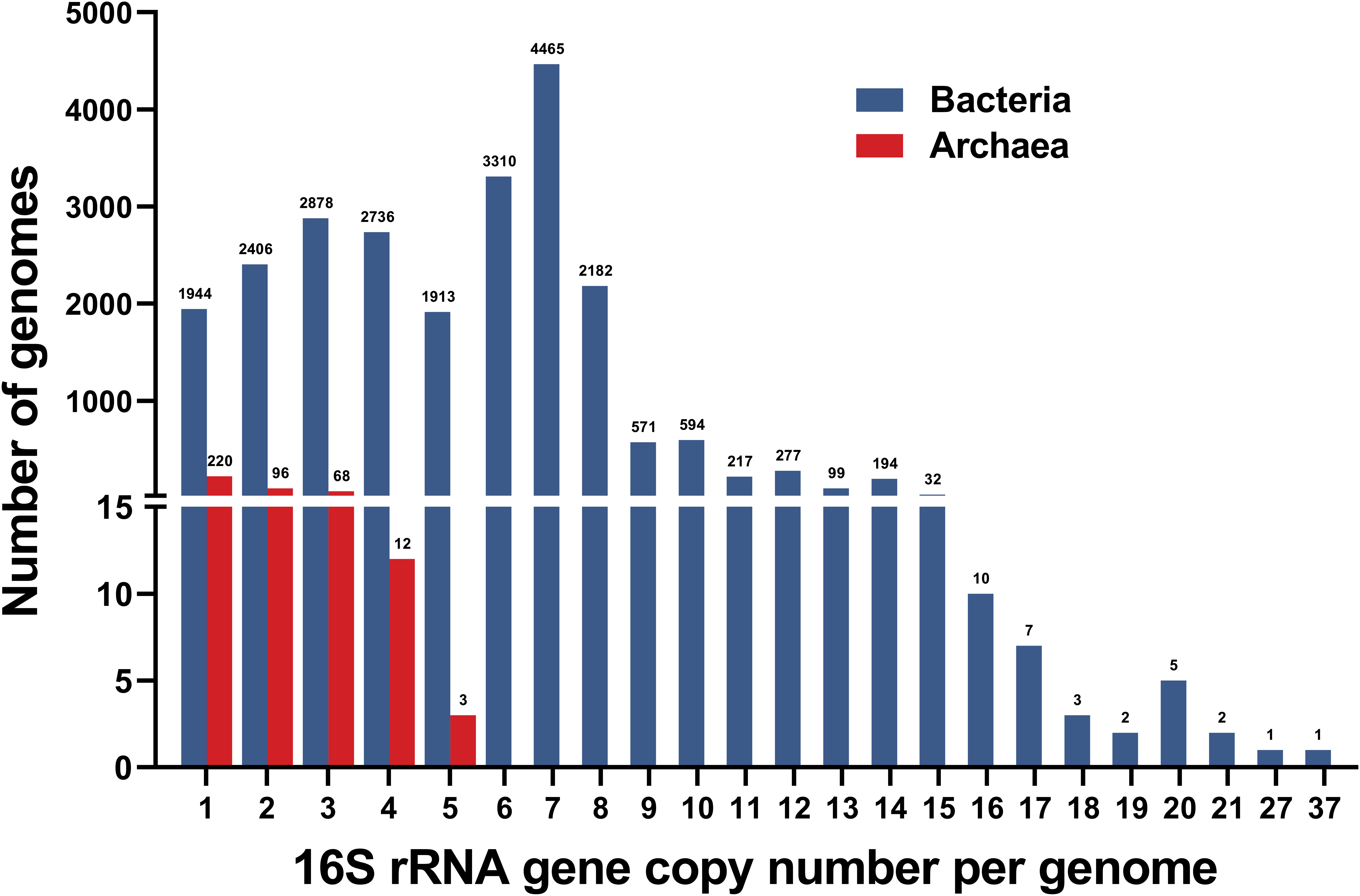
图1:细菌和古细菌基因组中16S rRNA基因拷贝数分布
上面列的值表示包含相应的16S rRNA基因拷贝的基因组数量。
单个基因组中16S rRNA基因的拷贝数在多个分类水平上具有类群特异性。每个门的平均拷贝数在1-6.96(表1,图S1)。在细菌门中,厚壁菌门的平均拷贝数最高,为6.96,其次是变形菌门(5.56)和梭杆菌门(5.26)。酸杆菌门(1.16)、热脱硫杆菌门(1.16)和绿弯菌门(1.46)的平均拷贝数较低。对于古菌门,16S rRNA基因的平均拷贝数在广古菌门中为2.06,在奇古菌门中为1.26,而其余古菌门平均只含有1个拷贝的16S rRNA基因。
原核生物16S rRNA基因组内异质性
许多原核生物基因组中含有多个16S rRNA基因,作者进一步研究了不同拷贝是否相同以及可能存在的差异程度。随着个体基因组内拷贝数的增加,16S rRNA基因序列变异体的数量呈上升趋势(图2A)。平均而言,一个基因组中存在三种16S rRNA基因变异体。在约60%的基因组(14502/24248个基因组)中观察到16S rRNA基因序列的基因组内变异(图S2),其中变异体数量在2-37之间。每个基因组内检测到的基因组内变异的详细信息可在File S1中找到。检测到的异质性在大部分DNA序列中低于1%(12642/14502个基因组),但其余1860个基因组的基因组内异质性大于1%,这可能会导致基于16S rRNA方法的错误分类。在Tumebacillus avium AR23208中有多达37个变体,并且变体之间的距离从0.06%到1.49%不等。计算出的最大异质性发现在Listeria monocytogenes 10-092876-1155 LM6中(27.9%)(图2B),其中包含5个不同的16S rRNA基因拷贝(图S3)。值得注意的是,尽管古菌通常含有较少的16S rRNA基因拷贝,但在具有多个拷贝的179个古菌株中,有119个存在基因组内差异。此外还发现,某些物种中具有两个或更多测序基因组的所有菌株都表现出高基因组内变异,如表S1所示,包括致病菌Borreliella afzelii和Streptococcus iniae以及极端微生物Thermopisa bispora、Haloarcula Marismortui、Haloarcula hispanica和Halomicrobium mukohataei。这些物种中稳定存在的不同rRNA变异体表明变异体可能具有特定的生理功能。
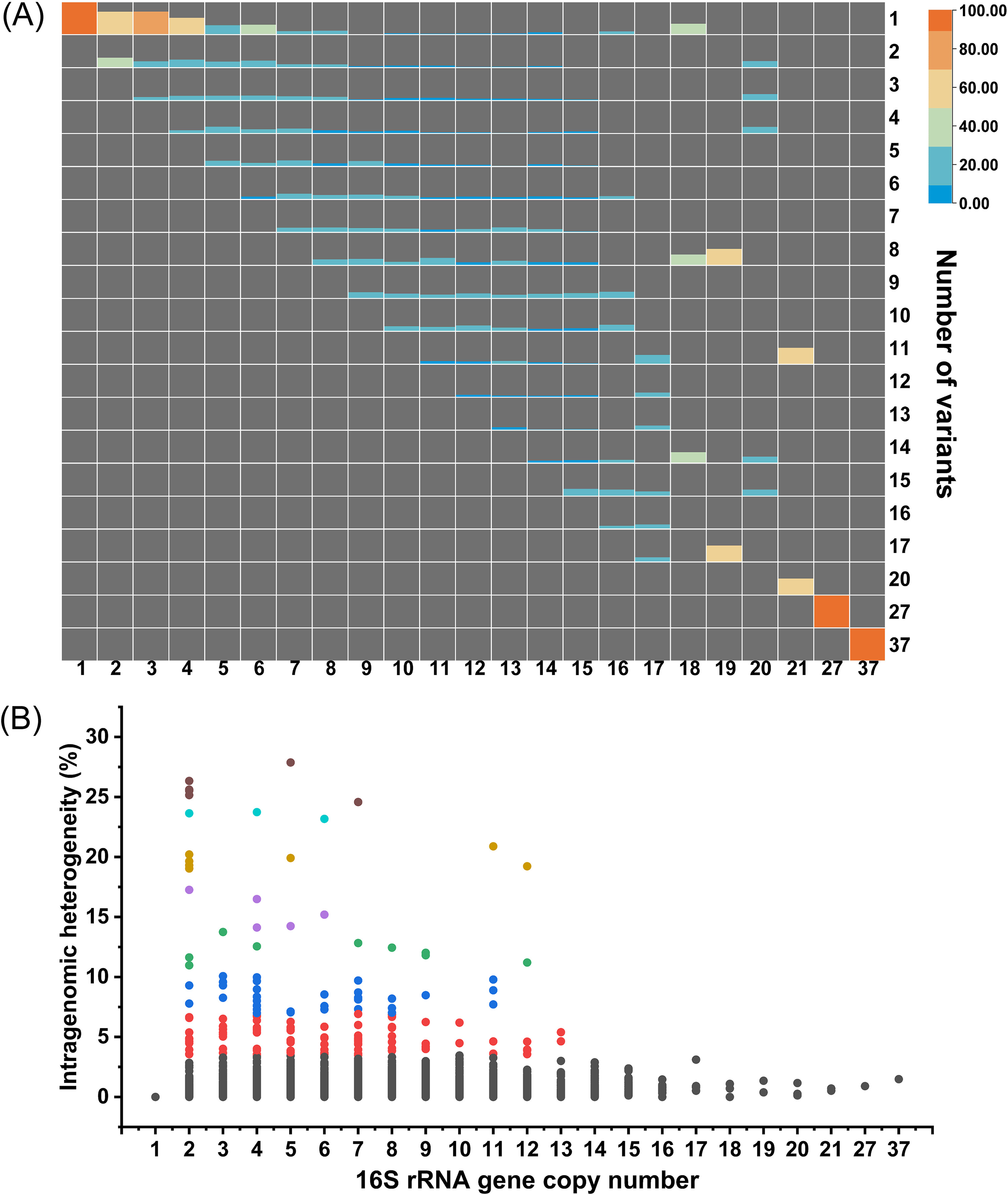
图2:不同16S rRNA基因拷贝数的原核生物基因组中变异数量及变异序列差异
(A) 不同16S rRNA基因拷贝数的原核生物基因组序列变异数的百分位数分布。每个网格中的彩色区域表示包含相应16S基因变异数(纵坐标)的基因组数量占所有包含相应16S基因拷贝数(横坐标)的基因组数量的百分比。网格的底色为灰色,表示比例为0。
(B) 不同16S rRNA基因拷贝数的原核生物基因组的基因组内异质性;每个点代表一个基因组。
使用不同的高变区估计微生物多样性的偏差
由于16S rRNA基因的基因组内差异,多个变异体很可能被划分到不同的序列簇中,导致基于16S rRNA基因的方法高估了微生物多样性。在ASV水平,即阈值100 %,使用全长16S rRNA基因时高估了156.5 %,显著高于关注部分区域时的(27.3 % ~ 110.5 %)值。V6区的基因组内变异导致了最低的高估(27.3 %),其次是V4-V5区(28.3 %)和V4区(28.8 %)。在常用的97%阈值下,V4 ~ V5(4.4 %)和V7 ~ V9(4.4 %)区域的高估程度最低,而V6(14.0 %)和V1 ~ V2(11.7 %)区域的高估程度最高。当使用97%和100%之间的其他聚类阈值时也得到了类似的结果(图3)。值得注意的是,对于全长16S基因,基因组内异质性导致的物种丰富度高估在97%的阈值水平上比100%显著降低,这表明应该选择特定的阈值以避免对物种多样性的有偏估计。
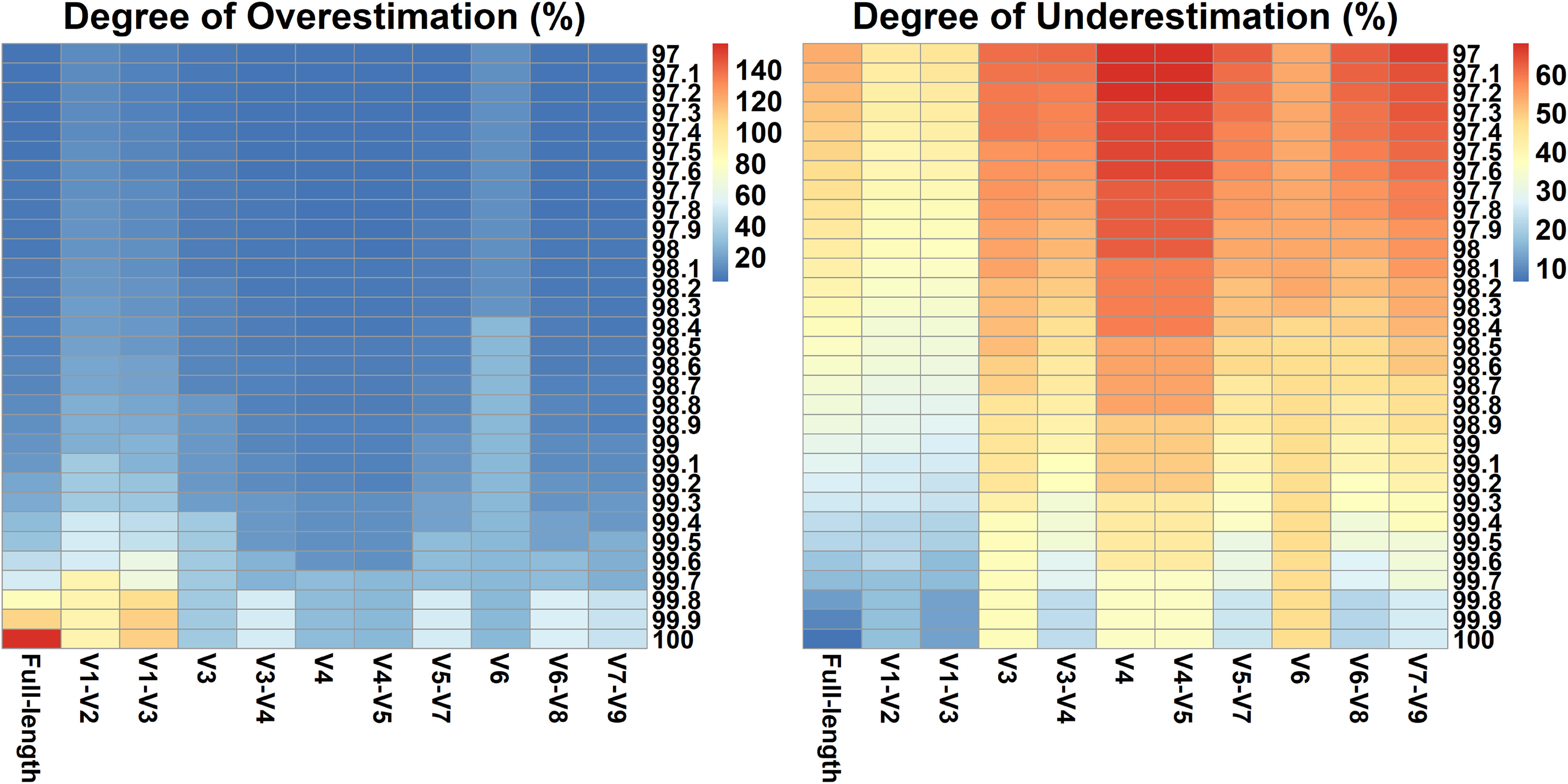
图3:全长和部分16S rRNA基因(横坐标)在97% - 100%的同一性阈值(纵坐标)下,基因组内异质性引起的高估程度(左图)和种间变异不足引起的低估程度(右图)。上述高估率和低估率是使用基于NCBI分类法构建的数据集计算的。如图例所示,从蓝色到红色的颜色范围分别代表从低到高的高估/低估率。
另一方面,一些亲缘关系较近但不同的类群可能共享相同或部分16S rRNA基因,即种间变异不足,导致微生物多样性被低估。通过比较聚类产生的物种数和OTU数来计算低估程度。对于较长的扩增子,如针对整个基因或V1-V3区的片段,在100%的阈值下,其多样性通常被低估了15%以下,而较短的扩增子,如V3、V4和V6区(表2 ,图3),则大大低估了物种数量。这些结果证明了一些由于某些类群中可变区的保守部分,近缘物种在使用部分16S基因时无法区分。
考虑到GTDB分类法是系统发育一致性和秩标准化的基于基因组的分类法,利用GTDB-Tk(v2)获得每个基因组的GTDB分类法等级,并构建16S rRNA基因序列集,计算基于GTDB分类法估算微生物多样性的偏差。显然,使用GTDB数据集相对于NCBI数据集(图S4;具体偏差数据见S1文件)得到了类似的结果。16S rRNA基因V4-V5区的基因组内异质性导致物种多样性(3.7% ~ 26.4%)在97% ~ 100%的阈值下被低估(图S4A)。如图S4B所示,全长扩增子在100%的一致性阈值下得到了接近物种数的多样性估计,仅减少1.7%的OTU。相比之下,部分基因扩增子导致OTU数量减少5%以上。这些结果表明,部分16S rRNA来自不同GTDB物种的基因倾向于聚集在一起,因此对进化关系的影响有限。
沿16S rRNA基因每个碱基的基因组内和基因组间变异
为了进一步研究可能导致物种多样性高估的潜在核苷酸位置,作者计算了古菌和细菌在16S rRNA基因上每个碱基的平均Shannon熵。熵用来表示每个碱基位置的基因组内变异,高熵值意味着核苷酸位置包含丰富的信息,也称为高度异质性。从图4A中可以看出,具有高Shannon熵值(0.04)的核苷酸位置倾向于集中在特定的区域,如V1、V3和V6区域,而V4、V5、V8和V9区域的核苷酸位点表现出低Shannon熵(0.02),这与观察到的靶向V4到V5区域引入的较小程度的高估相一致。相反,古菌不同可变区的熵值之间没有显著差异(图4B)。这可能是由于古菌基因组数量少于细菌基因组数量,另一种可能的解释是古菌基因组中通常含有较少的16S rRNA基因。在16S基因的每个核苷酸位置的基因组间变异方面,如图4C和D所示,在9个高变区中观察到了较高的Shannon熵。足够的基因组间变异的存在可能使基于16S基因片段的分析能够用于区分属和更高级别的不同类群。
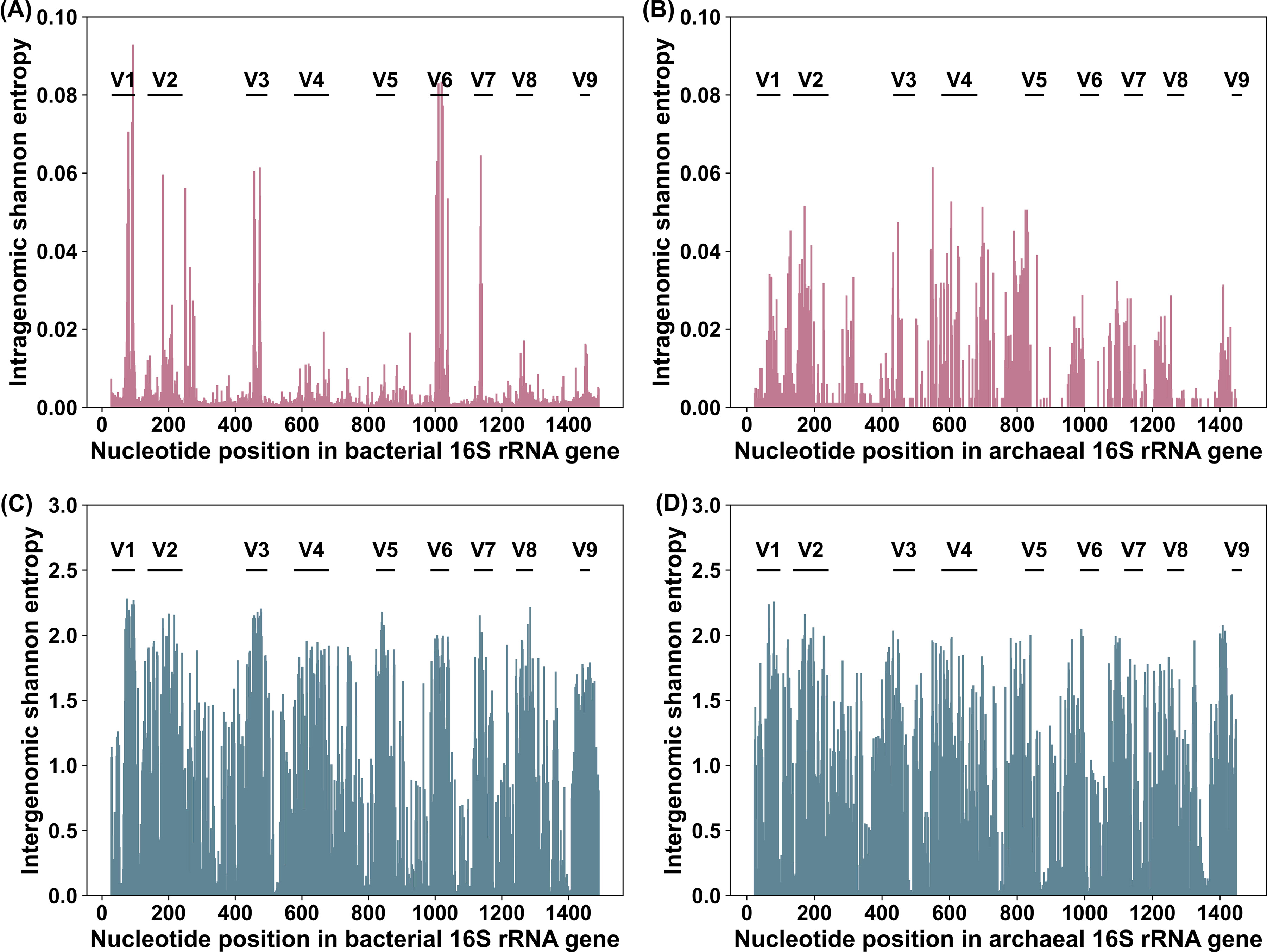
图4:细菌和古细菌16S rRNA基因的基因组内和基因组间变异
(A) 通过对所研究的所有23899个细菌基因组中16S rRNA基因每个位置的香农熵平均计算基因组内平均变异。序列坐标参照大肠杆菌K-12 MG1655的16S基因(基因ID 948332)。
(B) 在所研究的399个古菌基因组中,16S rRNA基因每个碱基的平均基因组内变异。序列坐标参照Saccharolobus solfataricus IFO 15331的16S基因(登录号NR_029127.1)。
(C和D) 基于SILVA Ref NR99数据库中所有细菌(C)和古细菌(D)序列比对的16S rRNA基因的基因组间变异。
不同聚类阈值下的过拆分和过合并率
对于11个扩增片段,如图5所示,在系列阈值下,计算了分配到多个聚类的物种百分比(过分裂)、包含来自不同物种序列的OTUs百分比(过合并)以及这两个值的总和(总误差)。总体而言,随着同一性阈值的增加,过分裂率也随之增加,当考虑全长rRNA基因时,阈值为100%时,过分裂率达到53%。当使用阈值为99%时,过拆分率均小于20%(图5)。说明较低的阈值可用于降低将同一物种内多个rRNA基因变异体拆分为不同OTU的风险。
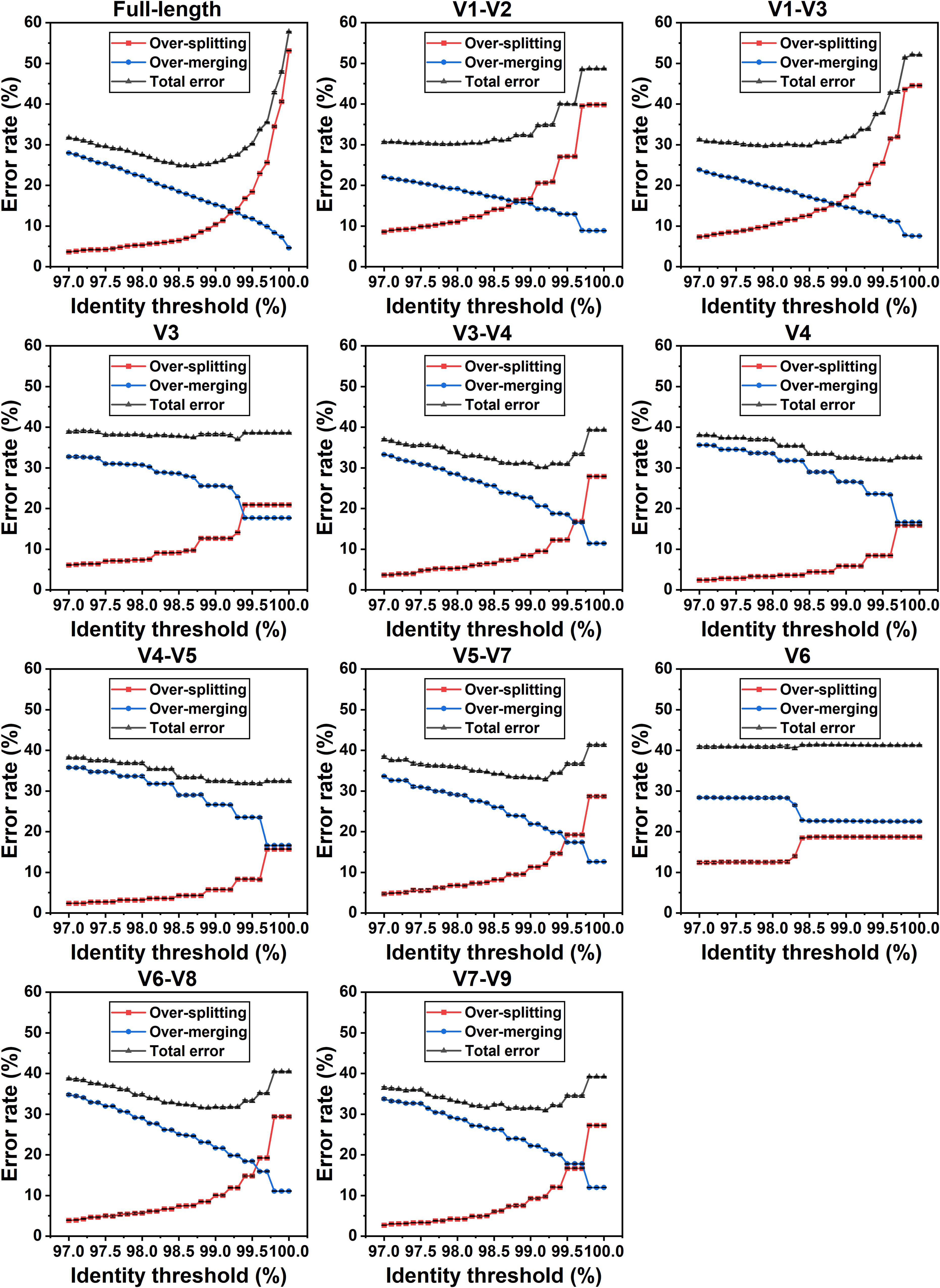
图5:聚类阈值为97% ~ 100%时,全长或部分16S基因的过裂、过合并和总错误率
使用基于NCBI分类法构建的数据集计算这三个指标。过度分裂率定义为物种被划分为多个聚类(OTU或ASV)的比例。淹没率计算为包含不同物种序列的簇的比率。总错误率表示上述两个值的总和。
与过拆分相反,随着聚类阈值的增加,过合并率表现出明显的下降。对于包含至少3个可变区的较长扩增片段,如全长区或V7-V9区,尽可能少;5%的重叠率出现在高阈值处。然而,对于包含一个或两个相邻可变区的较短片段,其百分比往往较高,例如V6区域高达22%。这表明当使用较短的扩增子时,来自不同物种的序列更有可能合并到同一个簇中。
如图5所示,当阈值从97%到100%变化时,总错误率有三种主要的变化趋势。一个是随着身份阈值的增加,如全长区域和V3-V4区域,有一个明显的先下降后上升的趋势,并且在特定阈值下可以找到最小值:全长16S rRNA基因的98.5%-99.0%,V1-V2区域的97.5%-98.0%,V1-V3区域的98.0%-98.5%,V3-V4区域的99.0%-99.5%,V6-V8区域的98.8%-99.3%,V5-V7 / V7-V9区域的98.7%-99.2%。另一种模式显示在V3和V6区域;在97%到100%的不同阈值下,总误差几乎保持不变。第三种趋势表现在V4和V4-V5区域;总误差随同一性阈值的增大而单调递减,两个变量区域的聚类阈值建议为99%-100%。此外,基于GTDB分类法构建的数据集(图S5)得到的最优身份阈值,即最小总错误率对应的阈值与基于NCBI分类法得到的阈值一致。
讨论
十年前的研究结果表明:古细菌每个基因组的16S基因拷贝数在1到4之间,细菌在1到15之间。由于数据库已扩增了近10倍,因此该结果需要被进一步分析。目前的分析显示,在现有的24248个原核生物基因组中,31个细菌基因组的16S rRNA基因拷贝数超过了之前的范围(15个拷贝),399个古细菌基因组中有3个超过了之前报道的最大拷贝数(4个拷贝)(图1)。不同菌株中16S基因的可变拷贝数极大地影响了每个分类群的相对丰度估计。例如,与拷贝数低的高丰度物种相比,可能更频繁地观察到拷贝数高的低丰度物种,这促使人们开发了试图克服这种偏见的工具,包括rrnDB、CopyRighter和PICRUSt。关于是否应通过平均16S rRNA基因拷贝数(GCN)校正相对丰度的问题一直存在争议。尽管文献中报告了GCN对微生物群落谱预测的改进,但最近的一项研究表明,16S GCN未能更可靠地揭示微生物生态学研究中的群落结构,这可能是由于参考数据库中拷贝数记录不足而受到限制。在这项研究中,作者提供了菌株水平或其他分类水平上更全面的拷贝数目录,这可以作为16S GCN的参考,以便更准确地估计微生物群落结构。
与之前的研究表明,在细菌和古菌中广泛观察到16S rRNA基因的基因组变异;这些菌株中有1860种含有多个拷贝的16S rRNA基因,其差异超过1%。此外,值得注意的是,所有菌株都表现出较高的基因组异质性(>1%)(表S1)的物种包括几种细菌物种,如热变形杆菌、爱德华氏菌属和纳氏紫单胞菌等,以及之前报告的一些嗜盐古生菌,如盐杆菌属、盐杆菌属和盐杆菌属。对于基因组异质性的出现,可能的解释是16S rRNA基因的水平基因转移(HGT),这已得到几项研究的支持。杨等人提供了证据,表明热变形杆菌染色体组的rrnB操纵子是通过水平基因转移从热变形杆菌或其他近亲物种获得的。有人提出,单一菌株中的rRNA基因分化是为了适应不断变化的自然环境,不同的变体在不同的环境条件下具有功能。例如,盐杆菌属携带了三个rRNA操纵子,其中操纵子B在核苷酸序列和GC含量上与其他两个操纵子明显不同,在高温(例如50°C)下比低温(例如15°C)下的表达水平更高。其他嗜盐古生菌也出现了不同rRNA基因受温度影响的表达现象,如盐杆菌属、盐杆菌属和盐杆菌属。此外,不同的rRNA变体在核糖体水平调控基因表达和细胞表型。在维布赖斯变形菌中,携带最多样化rRNAs的核糖体,即I-ribosomes,优先翻译与应激反应和碳代谢相关的特定mRNA,从而产生快速适应温度和营养变化的能力。作者对大量测序基因组进行的基因组异质性分析可以为进一步探索同一菌株内不同rRNA基因变体的起源和功能提供参考。
基因组内异质性是使用基于16S rRNA基因的分析来估计微生物多样性的干扰因素,可能会高估观察到的OTU数量。在ASV水平(100%阈值),全长或部分16S基因的高估率在27.3% ~ 156.5%之间(表2),普遍高于之前报道的数值。这可能是由于使用了更多的含有更高拷贝数16S rRNA基因的基因组。V3 ~ V4区因其整体覆盖度高、门谱广而成为微生物生态学研究中广泛使用的测序靶标;然而,基于V3 ~ V4区的扩增子分析可能导致微生物多样性高估51.7%。在最常用的阈值97%下,使用V4 ~ V5区域进行分析时,微生物多样性被高估了4.4%,是本研究中使用的所有片段中最低的。同时,16S rRNA基因的种间保守性可能会低估微生物的多样性,因为高度保守的近缘但不同物种的序列容易集中在一起。作者还发现V4 ~ V5区域表现出相对较高的低估率,OTU数量比物种数量低36.2%,即使使用最严格的阈值100 %,也表明物种分辨率较低。显然,如果希望在物种水平上实现分类学解析,则不建议将V4 ~ V5区作为测序目标,全长测序似乎更为合适。
序列聚类已常规用于从高通量测序平台检索到的扩增子读取的生物信息学分析,并根据给定的身份阈值将高度相似的序列分配给多个簇,这显著提高了下游分析的效率。事实上,聚类阈值的选择显著影响了群落α多样性的计算,应该仔细考虑。低阈值(例如97%)可能会将来自不同物种甚至属的序列聚类到相同的OTU(过度合并),因为属于同一属(例如埃希氏菌)的不同物种的16S rRNA基因在序列身份上高度相似,而阈值过高则可能由于基因组内变异而将物种甚至菌株分裂成多个不同的簇(过度分裂)。理想情况下,聚类过程产生一组OTU或ASV,每个OTU或ASV对应一个特定的分类单元,所有分类单元处于同一分类水平,以便于比较。此外,由于不同的可变区域提供了不同的分类分辨率,并且不同的扩增片段的适当阈值应单独确定,而不是取决于一般经验。因此,作者还尝试寻找全长和10个部分16S基因片段的物种区分的最佳鉴定阈值,以最小化总误差率。⭐⭐⭐对于全长16S基因,阈值为98.5%至99.0%时,总误差率相对较低(25% ~ 26%)(图5),而阈值超过99%时,总误差率迅速增加。对于V4或V4 ~ V5区域,阈值在99% ~ 100%之间时总误差最小;这意味着ASV比阈值为97%的OTU更适合采用。⭐⭐⭐
必须承认,无论采用哪个阈值,当仅使用16S rRNA基因作为测序目标时,都不可能获得实际的微生物结构组成。因此,我们所能做的就是在估计微生物多样性时尽量减少偏差。作者的研究结果为原核生物基因组中16S rRNA基因的异质性造成的高估程度和种间变异不足造成的低估程度提供了定量数据。作者还提出了全长和多个部分16S基因的最佳识别阈值,以最大限度地减少过聚类和过分裂的风险。有趣的是,当用GTDB分类法代替NCBI分类法进行上述分析时,关于估计微生物多样性的偏差以及不同测序区域的适当识别阈值的结论基本保持不变。该研究将有助于了解原核生物基因组中16S rRNA基因冗余和异质性如何影响微生物多样性的估计,并将为在微生物生态学研究中使用16S rRNA基因分析时选择扩增区域和聚类阈值提供一般指导。