Multi-genome metabolic modeling predicts functional inter-dependencies in the Arabidopsis root microbiome

摘要
背景:从理论生态学的角度来看,微生物群比预期的要复杂得多。除了竞争和竞争排斥之外,合作微生物之间的相互作用也必须仔细考虑。微生物之间的代谢依赖性可能解释了微生物群的共存。
方法:在这项计算机研究中,作者探索了从拟南芥根中分离的193种细菌的基因组尺度代谢模型(GEMs)。作者分析了它们在模拟营养限制下的可生产代谢物,包括“根分泌物模拟生长介质”,并评估了为避免这些限制而进行的副产物和最终产物的假定代谢交换的潜力。
结果:作者发现基因组编码的代谢潜能在数量上和质量上都是通过系统发育聚集的,突出了不同分类群之间的代谢分化。增加菌株数量的随机合成组合(SynComs)表明,随着平均系统发育距离的增加,GEMs可生产的化合物数量增加,但大多数SynComs都集中在最佳系统发育距离附近。此外,由于代谢冗余,相对较小的syncom可以反映整个群落的容量。对30种特定终产物代谢物(即目标代谢物:氨基酸、维生素、植物激素)的检测表明,大多数菌株具有产生几乎所有目标化合物的遗传潜力。它们的产生被预测为:(1)依赖于外部营养约束,(2)由模拟根分泌物的营养约束促进,这表明养分有效性和根分泌物在决定可生产代谢物的数量方面起着关键作用。一个答案集编程求解器能够识别出许多菌株的组合,这些菌株预计在严重的营养限制下相互依赖以产生这些目标化合物,从而表明假定的亚群落水平的功能冗余。
结论:这项研究预测了环境中可用营养物质引起的代谢限制。通过扩展,它强调了环境对利基潜力、实现、划分和重叠的重要性。作者的研究结果还表明,根微生物群成员之间的代谢依赖和合作弥补了环境的限制,并有助于维持复杂微生物群落的共存。
介绍
众所周知,微生物通过建立密集的相互作用网络并促进维持宿主稳态的基本功能来定植大型生物体。这些功能是多种多样的,从防止病原体到营养吸收,以及抵抗诸如高温或干旱等压力。与温度、pH值、氧气、营养物质或优先效应等其他因素一起,宿主本身就是一个影响其微生物群组成的生态位。在植物中,土壤可以被认为是微生物的储存库,微生物从土壤中被招募形成根微生物群,根分泌物在土壤中起着积极的作用。最后,微生物-微生物相互作用在塑造微生物群的结构和动力学中是必不可少的,从而形成密集的相互作用网络。
了解微生物之间的多种相互作用是实现对微生物群功能的整体,社区水平理解的关键一步。这些微生物之间的相互作用存在于从竞争到合作的范围内,这取决于时间、空间、其他物种的存在和能量成本等。有两种主要的相互竞争的理论解释微生物的组合。生态位分化理论指出,系统发育上相似的物种更有可能竞争,因为它们具有共同的功能特征和重叠的资源,从而导致不太可能共存。生境过滤理论认为优势种表现出相似的功能特征,因为它们的存在是由环境参数决定的。竞争与合作的相对重要性尚不清楚。不同的方法产生了相互矛盾的结果:几种计算方法预测交叉喂养的可能性,而几种体外实验则指向竞争Understanding Competition and Cooperation within the Mammalian Gut Microbiome 。然而,据报道,代谢相互作用,特别是代谢依赖性在维持群落多样性和稳定性以及解释微生物共存方面发挥着重要作用。因此,细胞外代谢物可以在微生物群落组装中发挥主要作用,菌株之间的代谢依赖性可以解释为什么一些微生物不能在标准实验室条件下培养。
微生物系统生态学现在经常用于模拟复杂的系统,如生态过程。组学数据的获取和分析,再加上建模方法,使得计算预测生物体在不同条件下的资源利用、生物合成能力、缺陷和生长成为可能,特别是可用营养物质,以下称为“营养约束”(表1)。这些模型依赖于代谢网络的重建,即来自注释基因组的基因组尺度代谢模型(GEMs)。因此,使预测基本生态位重叠和同一微生物群落成员之间的竞争成为可能。研究微生物群落的系统发育结构也可以检测系统发育信号和代谢之间的相关性。
作者在计算机上研究了系统发育如何在单个菌株(1)和菌株的小随机组合(SynComs)(2)的规模上塑造GEMs,测试了可用营养物质(包括根分泌物)对GEMs的约束作用的强度(3),并检验了菌株之间的代谢合作是否可以缓解这些营养约束(4)。作者测试了四个假设:
(i) 无约束代谢(见表1中的定义)通过系统发育高度聚集,这意味着根据菌株的分类,预测的可生产代谢物在菌株之间是分化或重叠的(H1);
(ii) 根据相应细菌之间的系统发育相似性,GEMs组合比单个GEMs具有更多的可生产代谢物(H2);
(iii) 可利用的营养物质可能对细菌的代谢产生影响,导致可生产代谢物从无约束代谢减少到约束代谢(H3);
(iv) 代谢合作是常见的,并且可能通过允许特定关键化合物的产生来补偿营养限制,这是在一个细菌产生的每种化合物都可以与其他细菌共享的强大假设下。这一假设至少在一定程度上得到了一些分析或预测细菌和植物分泌体的研究的支持(H4)。
为了验证这些假设,作者分析了从拟南芥(Arabidopsis thaliana)根部分离的细菌菌株的基因组,并使用系统生物学方法在计算机上预测基因组功能。利用基因组注释重建的GEMs预测了每种细菌的代谢。
表1 所使用指标的描述
在这里,术语“群落”是指整个群落或菌株的随机子样本(SynCom)。表格中加粗的首字母缩略词在全文中使用
| Metric | Description |
|---|---|
| Nutritional constraint | Available nutrients on which a GEM can rely on (i.e., the initial reactants of the whole network). Nutritional constraints are modeled with simulated growth media. In this paper, an “unconstrained” GEM represents its metabolic potential, i.e., all the metabolites it encodes and can theoretically produce. |
| Predicted Producible Metabolites (PPM) | The list (number and composition) of all metabolites predicted to be producible by one or several GEMs simultaneously (also referred to as a meta-GEM), under a nutritional constraint or in the absence of a constraint. This metric is used to summarize the unconstrained and constrained (by available nutrients) metabolism inferred from genomes. |
| Core Predicted Producible Metabolites (CPPM) | The part (number and composition) of a community PPM which is individually producible by each GEM individually in a set of GEMs. |
| Targeted Predicted Producible Metabolites (TPPM) | A set of 30 metabolites on which part of this study is focused. Their ability to be produced by one or several GEMs is analyzed (number and composition), under a nutritional constraint or in the absence of a constraint. |
| Community added value | The part of the PPM (number and composition) of several GEMs which is only producible by metabolic interactions within a community (i.e., not producible by a single GEM). |
| Average phylogenetic distance | The average of all pairwise phylogenetic distances between pairs of strains in a synthetic subsample of strains (SynCom). The whole community also has an average phylogenetic distance. |
材料和方法
表1列出了本研究中使用的具体词汇和定义。对于基因组序列数据处理和指标获取的总结,见图1。
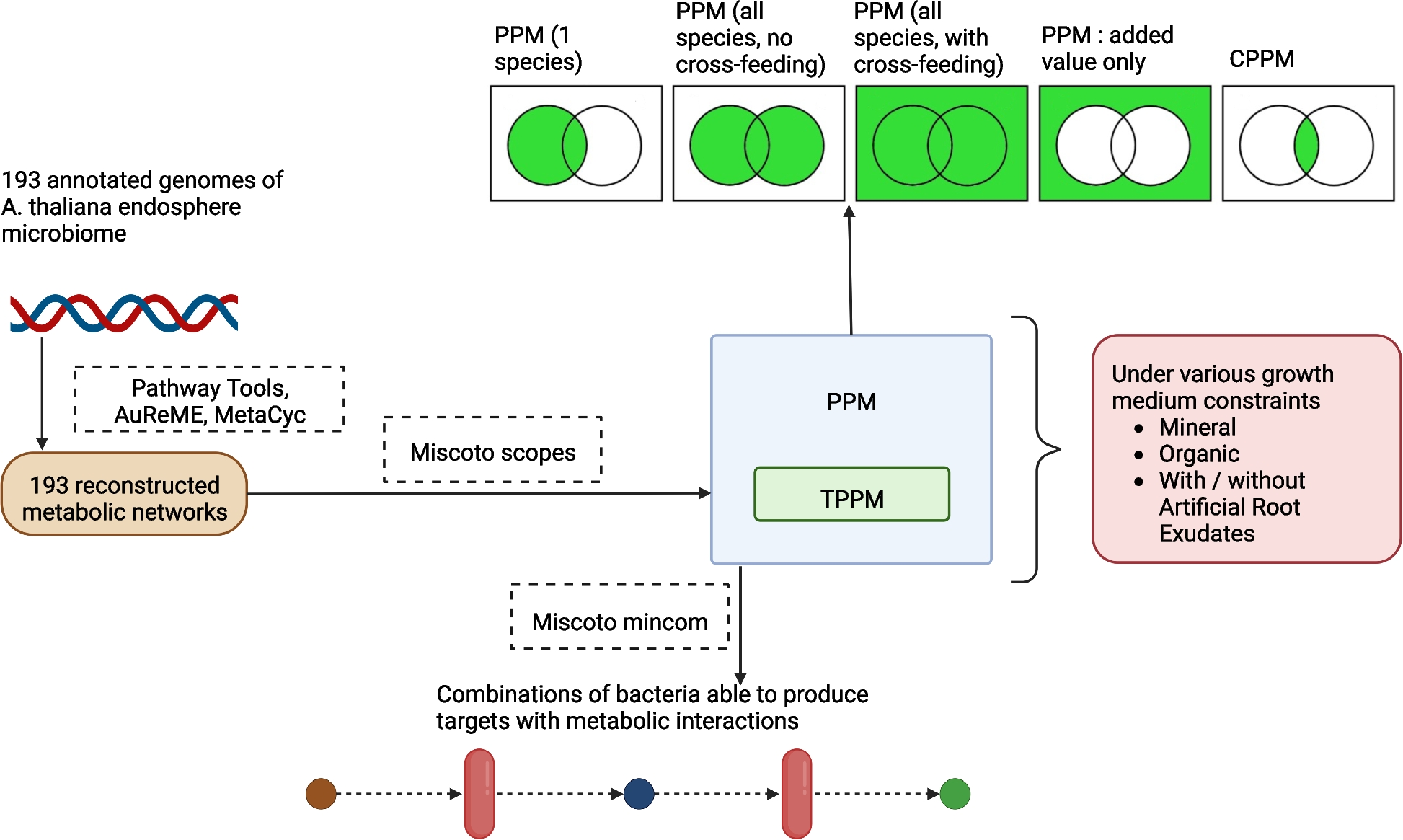
图1 基因组序列数据处理和度量获取。
作者的分析依赖于重建的代谢网络,每个基因组一个,在几种营养限制下计算PPM和TPPM的产量。PPM也可以为几个合作gem组成的社区计算:在这种情况下,仅由社区产生的PPM和TPPM部分(“附加价值”)也会返回。最后,计算能够产生尽可能多的TPPM的GEMs的最小组合
基因组数据
作者使用了193个带注释的基因组,这些基因组属于从生长在科隆农业土壤(德国)的拟南芥根中分离出来的细菌,代表了宿主植物的分类多样性核心细菌集。从At-SPHERE数据库(http://www.at-sphere.com/)下载带注释的基因组。同时使用了整个基因组的分类和系统发育。通过对31个细菌AMPHORA基因的多序列比对,将Clustal Omega v1.2.1传递到FastTree v2.1,通过最大似然推断出系统发育树。基因组在HiSeq2500上测序,被认为是高质量的草稿基因组(见Bai等人)。使用Trimmomatic v0.33对末端reads进行修剪。用A5和SOAPdenovo进行基因组组装。选择contigs最少的作为菌株的基因组组装结果。使用Prokka v1.11和SEED子系统,使用RAST服务器API进行功能标注。评估装配质量的数据(N50, N90,总长度,contigs数,reads数)可在Bai文章的补充数据3和4中查看。另外,图S1提供了基因组信息的图形摘要。作者还用Busco v5检查了基因组注释的质量。总体而言,在Busco数据库中注册的大部分核心基因都在基因组中被识别出来(即在几乎所有基因组中,超过95%的搜索到的Busco基因被预测出来,Supplementary Figure S1C)。
参考数据库
将基因组注释与代谢联系起来的参考是MetaCyc数据库,这是一个生物体特异性通路/基因组数据库(PGDBs)的集合。MetaCyc包含来自许多生物体的约2500种代谢途径。有两个标准促使作者选择这个数据库:首先,它是人工管理的;其次,作者为代谢网络重建设计的工具(mpwt和AuReMe)预先配置为与这个精心策划的数据库一起工作。
代谢网络重建(GEMs)
每个基因组的代谢网络使用基因组尺度代谢模型(GEMs)进行模拟,该模型使用自动命令行版本PathwayTools,使用metage2metabo工具套件的mpwt程序构建,然后使用AuReMe和padmet-utils将其转换为padmet和sbml格式。需要时使用Python lxml包解析sbml格式的GEMs。所有GEMs都是drafts,使用时没有空白填充或手工管理(这些步骤通常是提高GEM质量所必需的,但很可能引入假阳性,特别是在鲜为人知的生物体的情况下,从而掩盖了潜在的代谢依赖性)。因此,作者选择依赖GEM的drafts,也就是说,作者选择假阴性(由于基因组注释的缺陷)而不是假阳性。
基因组和GEM指标
为了检测菌株和代谢之间的模式,使用了一组指标(表1),并将其应用于单个GEM(即单菌株)和包含2-20个GEM的随机组合(SynComs)。Miscoto scopes的Python API用于计算模拟营养约束下单个GEM、SynComs和整个群落的所有预测可产代谢产物(PPM)。对AuReMe报告进行解析以记录无约束的PPM。使用Python包ete3基于系统发育树计算系统发育距离。基因组大小在注释数据中。对于每个SynCom,还使用Python 3、AuReMe和Miscoto作用域输出中的集合计算了核心预测可生产代谢物(CPPM)(图1)。还使用Python集合计算了整个社区的附加值。
目标预测可产代谢物(TPPM)
作者研究了单个GEM和整个社区(meta-GEM,其中所有GEM都可以泄漏和交换任何化合物)生产TPPM的生产能力。在模拟生长介质的各种营养限制条件下,GEM的TPPM产量可以计算出来(见下面的专门部分)。在这项研究中,作者关注的是30个TPPM:17种氨基酸(丝氨酸、丙氨酸和谷氨酸被排除在外,因为它们存在于人工根分泌物中,它们是生长介质的一部分),8种B族维生素(二磷酸硫胺素、核黄素、烟酸、(R)-泛酸、吡醇、生物素、四氢叶酸、腺苷钴胺)和5种植物激素(生长素、水杨酸、脱落酸、乙烯、茉莉酸)。选择氨基酸是因为它们具有基本的、无处不在的生物学重要性。维生素是根据它们在新陈代谢中的重要性来选择的。根据所研究微生物群落的根相关性状选择植物激素。此外,根据参考数据库,这些代谢物生物合成途径及其编码基因相对已知,易于预测,并且在所研究的分类群中是预期的,从而降低了基因组组装缺失、注释错误和重建代谢网络假阴性的风险。在营养限制下(见下文),使用Miscoto scope的Python API计算每个GEM的TPPM产量。在没有营养限制的情况下,在AuReMe报告中评估了TPPM的产生情况。
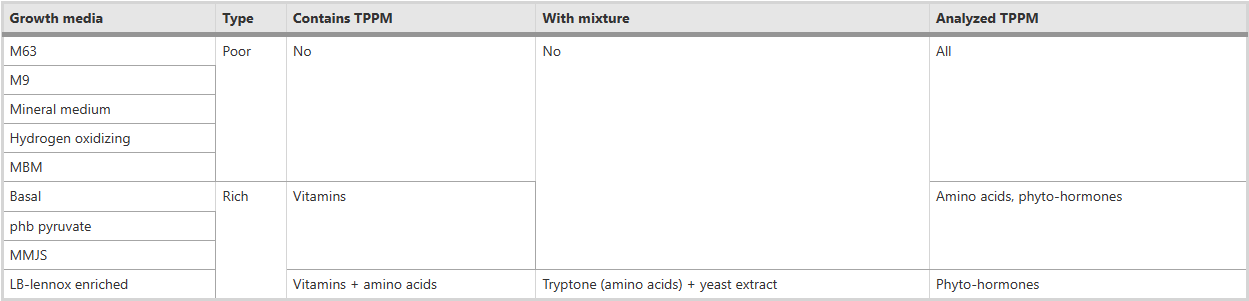
表2 用于营养约束的培养基总结
由于富培养基总是含有一些TPPM,因此所研究的生长培养基组因分析而异:TPPM要么一次分析,不含维生素,要么只分析植物激素,这取决于所涉及培养基的含量。有关培养基组成的详细信息可以在gitlab上找到
假定GEM组合的代谢相互作用
对于每种营养约束(即每种培养基),使用Miscoto mincom(在metage2metabo包中实现的一个版本),并将整个微生物群落作为输入(sbml格式的GEMs)。该工具使用答案集编程,这是一种面向组合问题解决的声明性方法。它迅速确定了在生长限制下能够产生尽可能多的指定TPPM的GEMs的所有最简单的组合(称为“解决方案”),复杂的解决方案(包含更多GEMs)将被忽略。TPPM要么是上述所有化合物,只有氨基酸,只有维生素,要么只有植物激素,符合上述条件“生长介质中没有TPPM”(表2)。分解TPPM也很重要,因为结果强烈依赖于TPPM:例如,具有生成给定TPPM的必要但罕见反应的GEMs将非常频繁地被表示出来,潜在地掩盖了其他TPPM的可能组合。每次运行的结果都以json格式存储。
准泊松广义线性模型(Quasi-Poisson GLMs)
在没有营养约束的情况下,计算随机SynCom的PPM/CPPM数量与SynCom大小、基因组大小和系统发育距离之间的相关性。首先,每个成员数(变量“SynCom大小”)在2到20之间(步长为1),通过从初始的193个菌株池中随机选择菌株构建500个SynCom,不进行替换。首先,评估解释变量之间的独立性(图S2D)。然后,基于准泊松分布(以对抗响应指标的过度分散,图S2, C1至C4),建立了两个广义线性模型(GLMs),根据平均系统发育距离、平均基因组大小和SynCom大小模拟了PPM和CPPM数量对SynCom的响应。补充系统发育距离多项式(2次)项,对响应指标曲线进行建模:Y∼P(μi,θ)
式中i为SynCom,Y为所含PPM或CPPM的数量,P为平均系统发育距离,G为平均基因组大小,S为菌株数(即GEMS)。模型中可包含的最大SynCom大小为12株。之所以选择这个大小,是因为它位于所有度量值或方差达到的平台的起点(图S2, B1至B4)。这也是当使用许多自举子样本进行测试时,较大的SynCom开始显示从一个SynCom大小到下一个SynCom大小没有显着差异的大小(对于每个SynCom大小:500次迭代,每个50个菌株,单侧Wilcoxon, Mann-Whitney检验,图S3)。由于不同大小的SynComs中值的分布不均匀,模型残差略有偏倚和非正态分布(图S4和S5)。由于作者使用的模型不会自动返回R^2^,因此它们使用以下公式计算:1 -残差/零偏差(可在模型的R摘要中获得)。该分析忽略了SynCom的PPM的附加价值,因为它是用Miscoto计算的,而Miscoto仅在应用的增长约束下工作。
其他统计分析
使用非参数检验(Wilcoxon和Mann-Whitney秩和检验)进行分类对公制分布的影响和培养基对PPM数量的影响的检验。用Cliff’s delta法计算相应的效应量。对Jaccard距离矩阵进行主坐标分析(PCoA),使用R包ape和Vegan的PCoA和vegdist函数。使用Adonis函数对距离矩阵进行PERMANOVA,该函数与Levene的方差齐性检验(PERMDISP2程序)的多变量模拟相关联,并使用相同包装的betadisper函数。根据相应的整个群落PPM组成(定性地,即哪些化合物在哪种营养约束下可以生产,通过多少GEMs)计算布雷-柯蒂斯距离(使用素食主义者函数),通过R基聚类函数(使用默认的“complete”方法)构建生长介质树状图。涉及分类学效应的测试排除了拟杆菌门和厚壁菌门,因为它们的样本量小(分别为4和7株),显著性阈值设为0.01。
脚本
使用自制的Python 3脚本组织数据采集和工具输入输出之间的链接(图1)。所有Miscoto输出都以json格式存储,其中包含的相关数据(PPM和TPPM,给定营养约束下产生TPPM的基因组数量等)被解析并存储为csv表。使用r4和ggplot2软件包以及Python 3和matplotlib和seaborn软件包进行图形和数据分析。脚本和数据可在https://gitlab.com/mataivic/article_metabolic_modelling_thaliana_microbiome上获得。