日期:2021年9月1日
参考:QIIME 2教程. 13训练特征分类器TrainingFeatureClassifiers(2021.2)_刘永鑫的博客——宏基因组公众号-CSDN博客
下载并导入参考序列
这里使用85_otus.fasta(按85%相似度聚类)的文件用于演示,是因为体积小运行更快。实际中一般使用参考序列99%和97%聚类的结果用于分类。如
# 创建工作目录
mkdir -p training-feature-classifiers
cd training-feature-classifiers
# 下载参考OTU数据集,7M
wget -c https://data.qiime2.org/2021.2/tutorials/training-feature-classifiers/85_otus.fasta
# 下载参考数据集的物种分类信息,442K
wget -c https://data.qiime2.org/2021.2/tutorials/training-feature-classifiers/85_otu_taxonomy.txt
# 下载代表性序列文件,1.7M
wget -c https://data.qiime2.org/2021.2/tutorials/training-feature-classifiers/rep-seqs.qza
将这些数据导入到QIIME 2对象中。由于Greengenes序列物种注释文件(85_otu_Taxonomy.txt)是一个不带标题的制表符分隔文件(tsv),因此必须指定HeaderlessTSVTaxonomyFormat作为源格式,因为默认源格式需要标题。
# 导入参考序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path 85_otus.fasta \
--output-path 85_otus.qza
# 导入物种分类信息
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path 85_otu_taxonomy.txt \
--output-path ref-taxonomy.qza
输出对象:
85_otus.qza: 按85%聚类的参考数据库。rep-seqs.qza: 预训练的参考数据库。ref-taxonomy.qza: 按85%聚类的参考数据库。
提取参考序列
2012年Werner等人研究表明,当一个朴素贝叶斯(Naive Bayes)分类器只训练被测序的目标序列的区域时,16S rRNA基因序列的分类准确度会提高。这种策略不一定对其他标记基因同样有效。从《4人体各部位微生物组分析》教程中知道,试图对序列进行分类的是120个碱基的单端序列,这些读长是用515F/806R引物对16S rRNA基因序列进行扩增的产物。我们在这里通过从参考数据库中提取基于与该对引物匹配的区域,然后将结果截取至120个碱基来对此进行优化。
# 按我们测序的引物来提取参考序列中的一段,1m
time qiime feature-classifier extract-reads \
--i-sequences 85_otus.qza \
--p-f-primer GTGCCAGCMGCCGCGGTAA \
--p-r-primer GGACTACHVGGGTWTCTAAT \
--p-trunc-len 120 \
--p-min-length 100 \
--p-max-length 400 \
--o-reads ref-seqs.qza
输出对象:
ref-seqs.qza: 提取的扩增区域。
注释:-p-trunc-len参数只能用于比对序列被剪裁成相同长度或更短的长度时,才需要剪裁参考序列。成功双端合并序列的长度通常是可变的。未在特定长度截断的单端读取的长度也可能是可变的。对于双端和未经修剪的单端读的物种分类,建议在适当的引物位置提取但不修剪为等长的序列进行分类器训练。
注释:用于提取扩增区域的引物序列应该是包含在引物结构中的实际DNA结合(即生物)序列。它不应包含任何非生物、非匹配的序列,例如接头、连接物或条形码序列。如果不确定引物序列的哪个部分是真正的DNA结合区域,咨询构建测序文库的工程师和测序中心。如果引物序列长度大于30nt,它们很可能包含一些非生物序列。
注释:上面的示例命令使用min-length和max-length参数,来排除使用这些引物远远超出预期长度分布的模拟扩增结果。这样的扩增产物可能是非特异性扩增,应该排除。如果将此命令调整为自己的项目中使用,请确保选择适合标记基因,而不是此处使用序列或参数。min-length参数在trim-left和trunc-len参数之后应用,在max-length之前应用,因此一定要设置适当的设置,以防止有效序列被过滤掉。
训练分类集
# 基于筛选的指定区段,生成实验特异的分类集,18s
time qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads ref-seqs.qza \
--i-reference-taxonomy ref-taxonomy.qza \
--o-classifier classifier.qza
输出对象:
classifier.qza: 生成分类器文件。
测试分类集
# 使用训练后的分类集对结果进行注释, 21s
time qiime feature-classifier classify-sklearn \
--i-classifier classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# 可视化注释的结果, 6s
time qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
输出对象:
taxonomy.qza: 生成物种注释结果。
输出可视化:
taxonomy.qzv: 生成物种注释可视化网页结果。
不进行训练的注释分析
time qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads 85_otus.qza \
--i-reference-taxonomy ref-taxonomy.qza \
--o-classifier classifier0.qza
time qiime feature-classifier classify-sklearn \
--i-classifier classifier0.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy0.qza
time qiime metadata tabulate \
--m-input-file taxonomy0.qza \
--o-visualization taxonomy0.qzv
对比
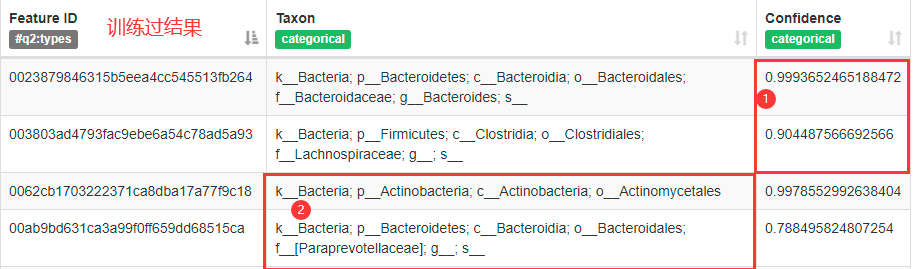
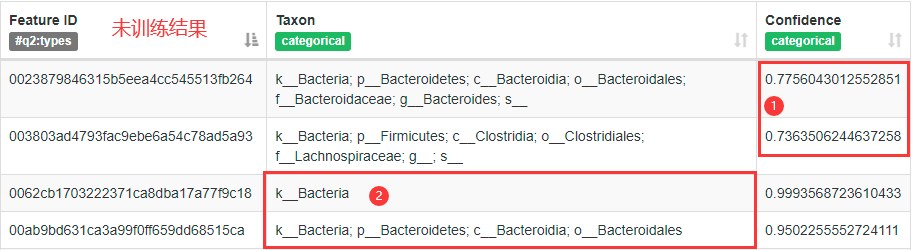
可以看到经过训练后注释的结果与未训练相比有以下两个优点:
- 注释的可信度更高(Confidence)
- 注释更详细(Taxon更具体)
结论
对于双端和未经修剪的单端读的物种分类,需要在适当的引物位置提取但不修剪为等长的序列进行分类器训练。