什么是K-mer?
mer 在分子生物学领域中意义为:monomeric unit,意即单体单元,相当于nt或者bp都可以。通常用于双链核酸中的单位,100 mer DNA相当于每一条链有100nt,那么整条链就是100bp。
k-mer 是指将reads分成包含k个碱基的字符串,一般长短为m的reads可以分成m-k+1个k-mers.
以k=40为例,kmer很容易按属水平分开细菌
k-mer是宏基因组序列拼接中DBG算法中的概念
通过把reads打断成一定长度的k-mer,然后根据k-mer之间严格的碱基配对关系构建de Bruijn图,最后通过对图形的解读找出最合理的序组装结果。
k-mer 如何影响宏基因组组装 ?
主要影响组装的特异性与敏感性
过短的k-mer不利于物种序列的区分,太长的k-mer不利于对拼接错误的矫正。
- 在测序深度足够的情况下,尽量选取长度较长的k-mer
- 在样本复杂度不高的情况下,尽量选取长度较长k-mer(其实原理与第一点一样)
- 利用一些新型的组装方法,如MEGAHIT这种选取不同长度k-mer进行迭代的方法,能有效同时兼顾准确性与组装长度。
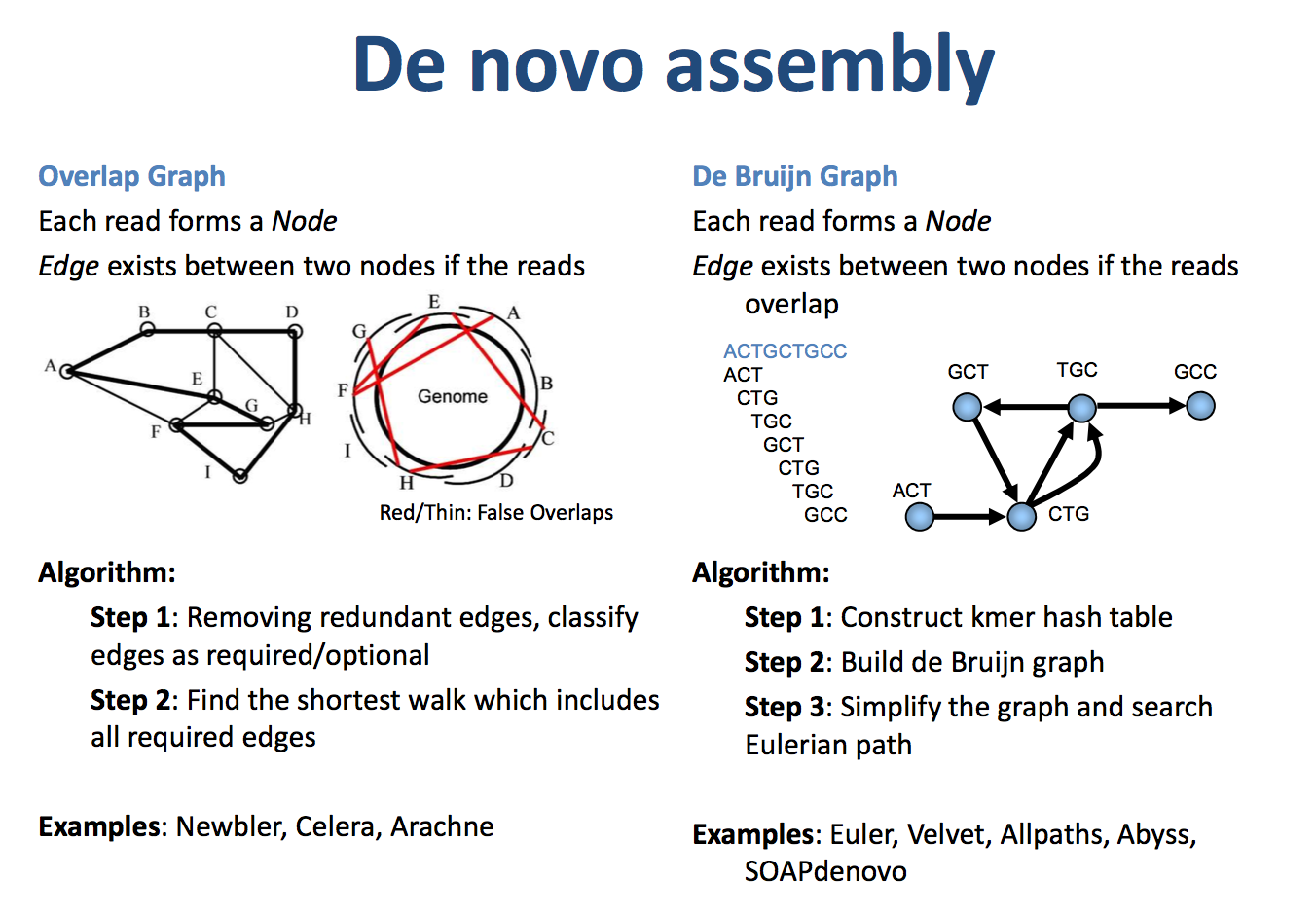
序列拼接的两种算法为OLC和DBG
overlap–layout–consensus (OLC)
OLC generally works in three steps: first overlaps (O) among all the reads are found, then it carries out a layout (L) of all the reads and overlaps information on a graph and finally the consensus © sequence is inferred 它是intuitionistic assembly algorithm,由Staden (1980)发明,主要用于长的低丰度序列的拼接,特别是一代数据,常见的软件有Arachne, Celera Assembler, CAP3, PCAP, Phrap, Phusion and Newbler.
de-bruijn-graph (DBG)
DBG是anti-intuition algorithm,它是首先将reads分割为更短的k-mers,然后来构图。1995 by Ramana M.Idury and Michael S. Waterman. 适合搞丰度的短的序列的拼接。相应的软件有Euler-USR, Velvet, ABySS, AllPath-LG, SOAPdenovo。争议一直存在,但软件的改进也没有停止过。
由于现阶段的主流测序方法是二代短片段测序,序列短而且数目庞大,如果利用overlap关系直接进行组装,这要求每对reads之间都进行一次序列比较,这会很耗费时间,而且结果并不可靠。为迎合二代测序的特点,一种基于k-mer的de Bruijn组装策略则成为更有效的解决方法。
经过预处理后得到 Clean Data,使用 SOAP denovo(肠道样品用soapdenovo soil,water用MEGAHIT)组装软件进行组装分析( Assembly Analysis )
if (micro diversity is not a major issue&& the primary research goal is to bin && reconstruct representative bacterial genomes from a given environment)
{
metaSPAdes should clearly be the assembler of choice.
# This assembler yields the best contig size statistics while capturing a high degree of community diversity, even at high complexity and low read coverage;
}elif(micro diversity is however an issue || the degree of captured diversity is far more important than contig lengths)
{
then IDBA-UD or Megahit should be preferred.
# The sensitivity of these assemblers, both for diversity as well as micro diversity, makes them optimal choices when trying to discover novel species in complex habitats. Whenever computational resources become limiting,
Megahit becomes the most attractive option, due to its good compromise between contig size statistics, captured diversity and required memory.
}
However, the bias of Megahit towards relatively low coverage genomes may provide a disadvantage for very large datasets, leading to a suboptimal assembly of high abundant community member genomes.
In such cases, Megahit may provide better results when assembling subsets of the sequencing data in a “divide and conquer” approach.
参考:
进一步探究: